

Saturated phosphatidic acids mediate saturated fatty acid-induced vascular calcification and lipotoxicity
- PMID: 26517697
- PMCID: PMC4665795
- DOI: 10.1172/JCI82871
Saturated phosphatidic acids mediate saturated fatty acid-induced vascular calcification and lipotoxicity
Abstract
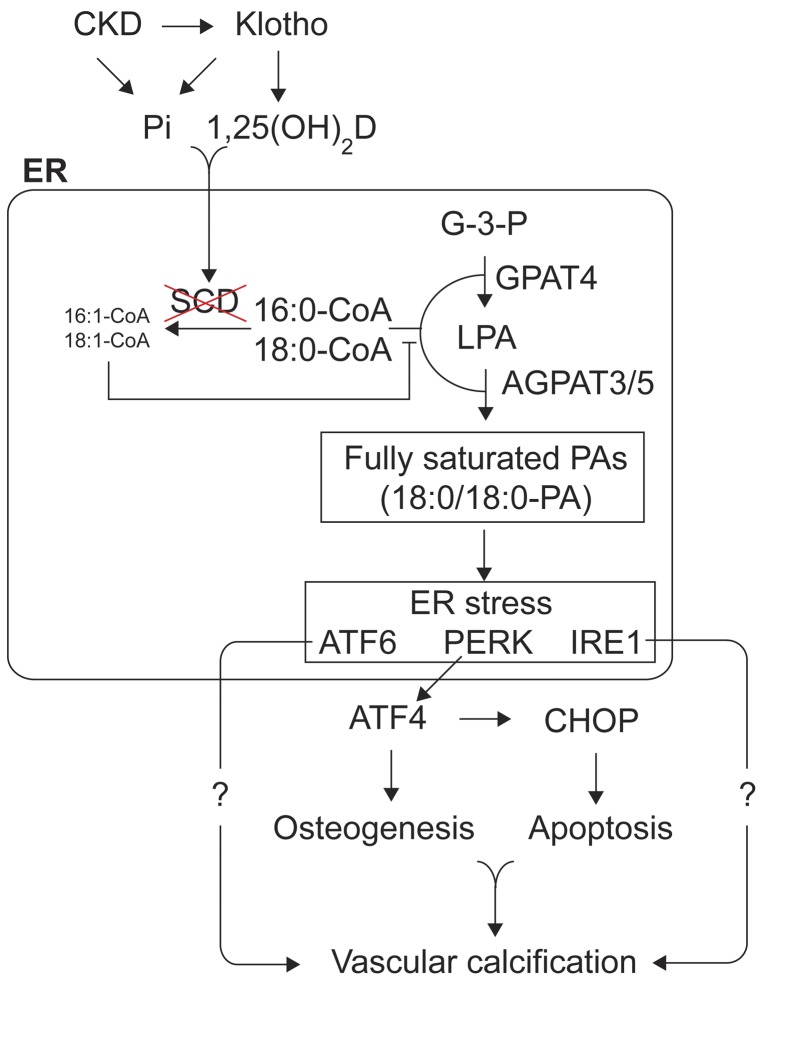
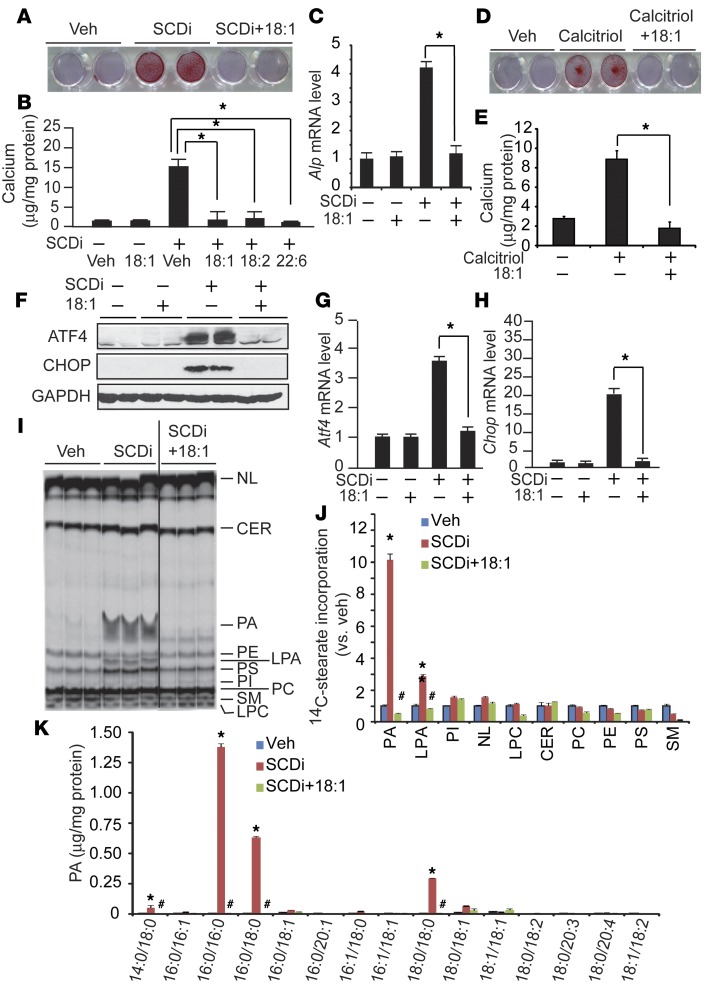
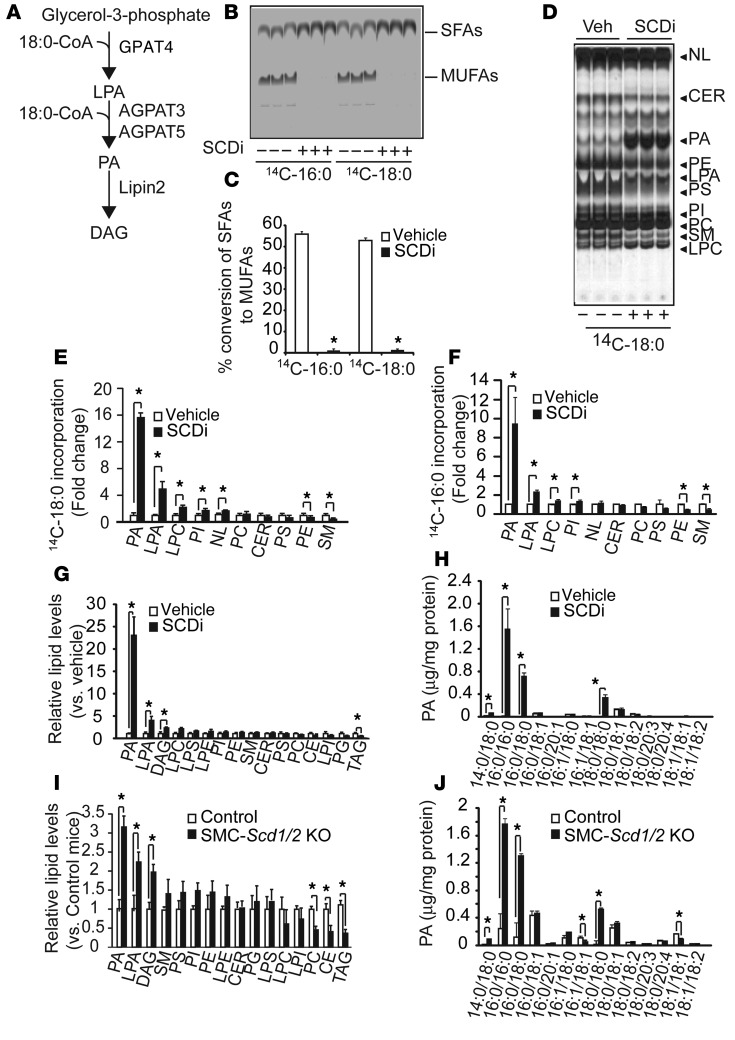
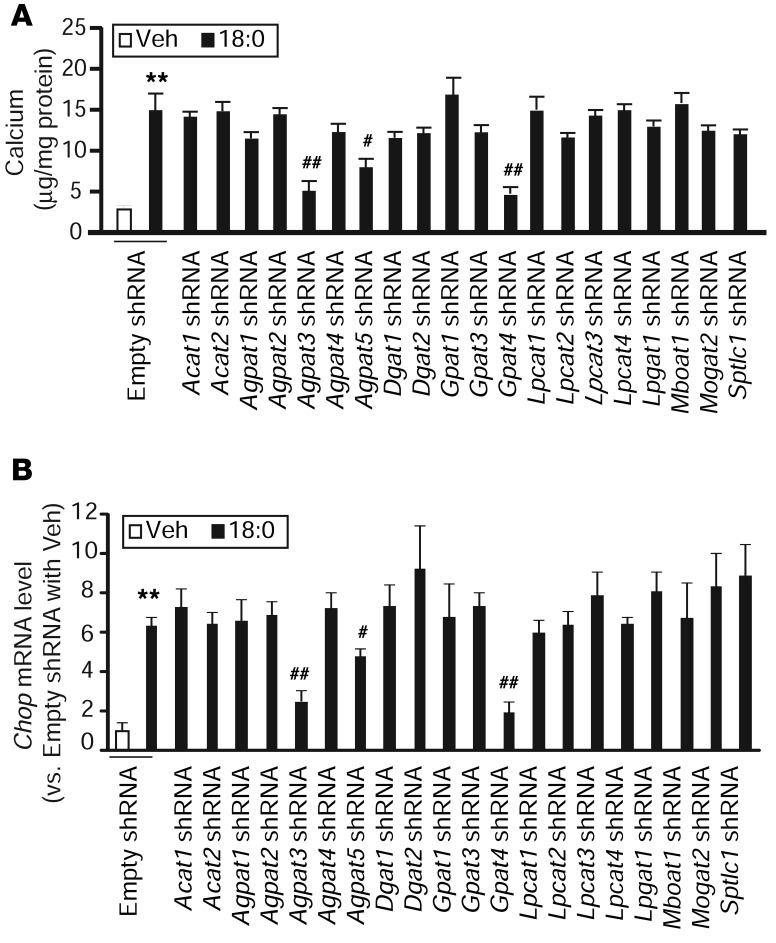
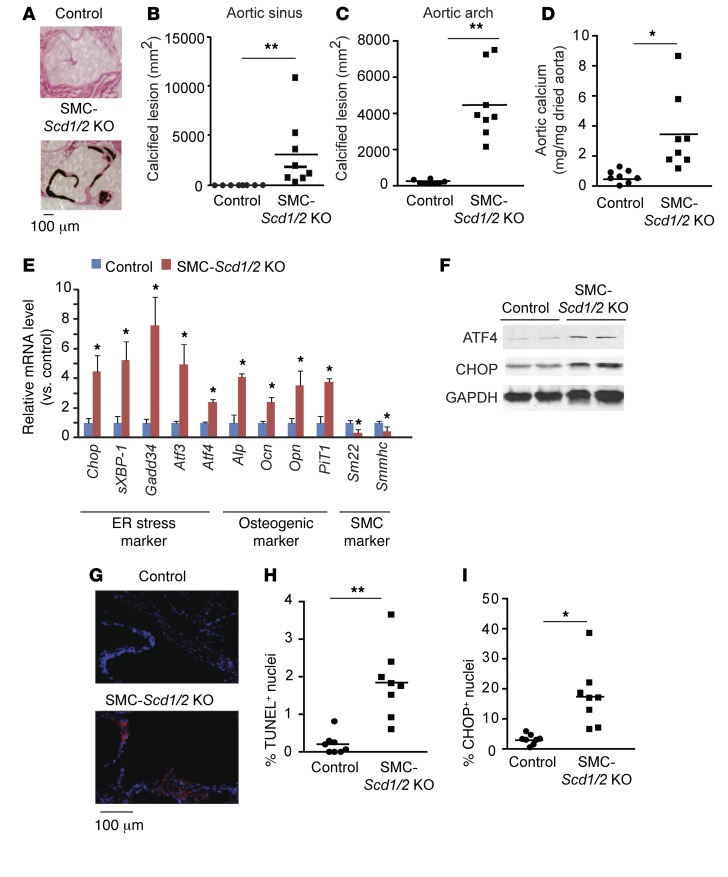
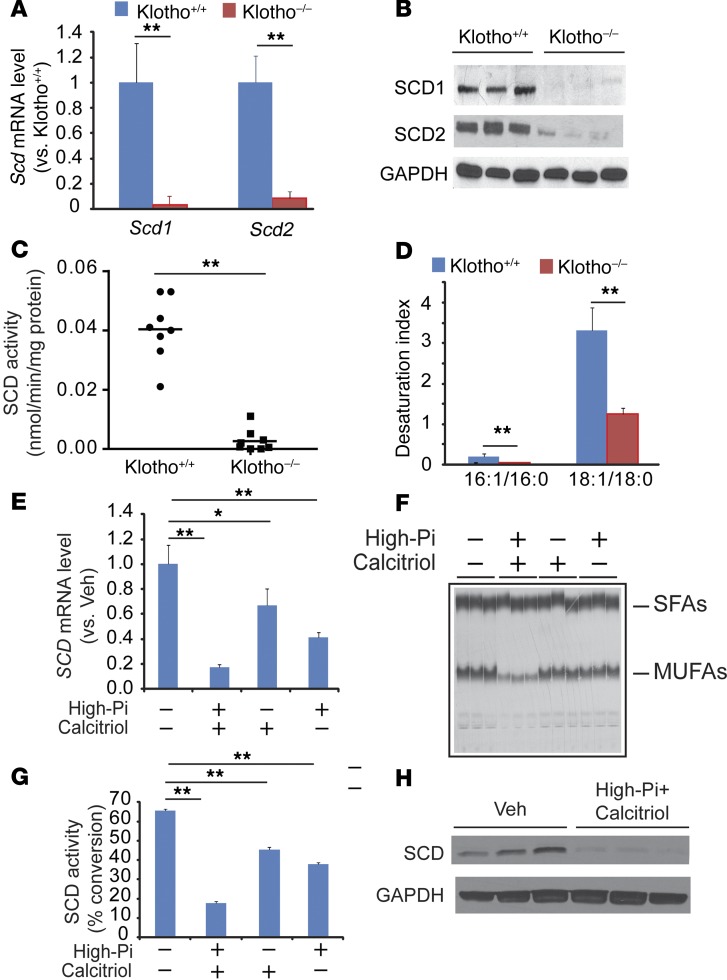
Recent evidence indicates that saturated fatty acid-induced (SFA-induced) lipotoxicity contributes to the pathogenesis of cardiovascular and metabolic diseases; however, the molecular mechanisms that underlie SFA-induced lipotoxicity remain unclear. Here, we have shown that repression of stearoyl-CoA desaturase (SCD) enzymes, which regulate the intracellular balance of SFAs and unsaturated FAs, and the subsequent accumulation of SFAs in vascular smooth muscle cells (VSMCs), are characteristic events in the development of vascular calcification. We evaluated whether SMC-specific inhibition of SCD and the resulting SFA accumulation plays a causative role in the pathogenesis of vascular calcification and generated mice with SMC-specific deletion of both Scd1 and Scd2. Mice lacking both SCD1 and SCD2 in SMCs displayed severe vascular calcification with increased ER stress. Moreover, we employed shRNA library screening and radiolabeling approaches, as well as in vitro and in vivo lipidomic analysis, and determined that fully saturated phosphatidic acids such as 1,2-distearoyl-PA (18:0/18:0-PA) mediate SFA-induced lipotoxicity and vascular calcification. Together, these results identify a key lipogenic pathway in SMCs that mediates vascular calcification.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
