

Inflammation and intracellular metabolism: new targets in OA
- PMID: 26521729
- PMCID: PMC4668929
- DOI: 10.1016/j.joca.2014.12.016
Inflammation and intracellular metabolism: new targets in OA
Abstract
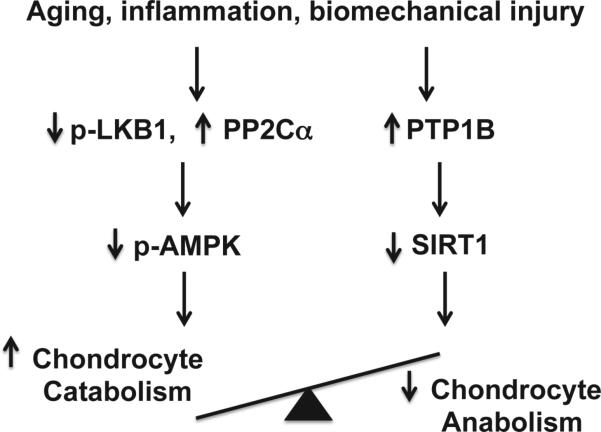
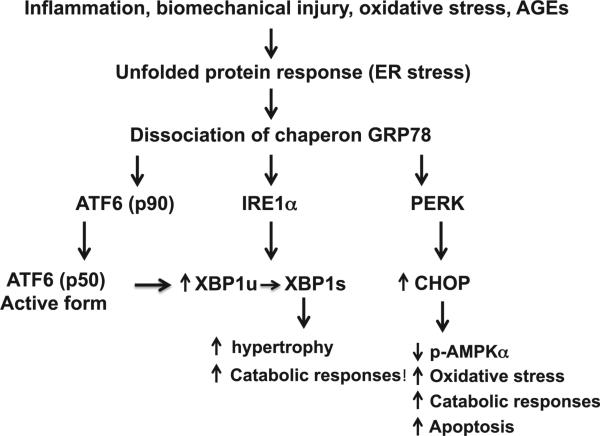
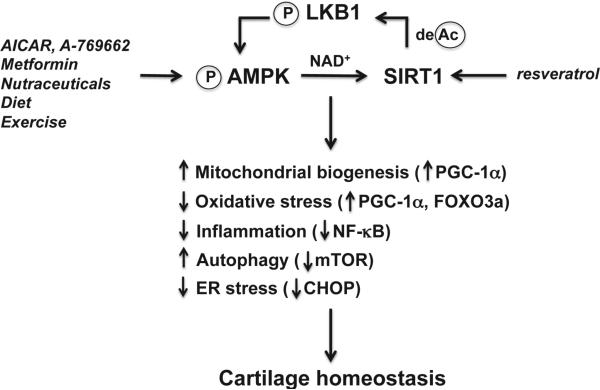
Articular cartilage degeneration is hallmark of osteoarthritis (OA). Low-grade chronic inflammation in the joint can promote OA progression. Emerging evidence indicates that bioenergy sensors couple metabolism with inflammation to switch physiological and clinical phenotypes. Changes in cellular bioenergy metabolism can reprogram inflammatory responses, and inflammation can disturb cellular energy balance and increase cell stress. AMP-activated protein kinase (AMPK) and sirtuin 1 (SIRT1) are two critical bioenergy sensors that regulate energy balance at both cellular and whole-body levels. Dysregulation of AMPK and SIRT1 has been implicated in diverse human diseases and aging. This review reveals recent findings on the role of AMPK and SIRT1 in joint tissue homeostasis and OA, with a focus on how AMPK and SIRT1 in articular chondrocytes modulate intracellular energy metabolism during stress responses (e.g., inflammatory responses) and how these changes dictate specific effector functions, and discusses translational significance of AMPK and SIRT1 as new therapeutic targets for OA.
Keywords: AMPK; Cartilage homeostasis; Energy metabolism; Inflammation; SIRT1.
Published by Elsevier Ltd.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
