

Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis
- PMID: 26523775
- PMCID: PMC4666835
- DOI: 10.1038/ng.3431
Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis
Abstract
Heritability analyses of genome-wide association study (GWAS) cohorts have yielded important insights into complex disease architecture, and increasing sample sizes hold the promise of further discoveries. Here we analyze the genetic architectures of schizophrenia in 49,806 samples from the PGC and nine complex diseases in 54,734 samples from the GERA cohort. For schizophrenia, we infer an overwhelmingly polygenic disease architecture in which ≥71% of 1-Mb genomic regions harbor ≥1 variant influencing schizophrenia risk. We also observe significant enrichment of heritability in GC-rich regions and in higher-frequency SNPs for both schizophrenia and GERA diseases. In bivariate analyses, we observe significant genetic correlations (ranging from 0.18 to 0.85) for several pairs of GERA diseases; genetic correlations were on average 1.3 tunes stronger than the correlations of overall disease liabilities. To accomplish these analyses, we developed a fast algorithm for multicomponent, multi-trait variance-components analysis that overcomes prior computational barriers that made such analyses intractable at this scale.
Figures
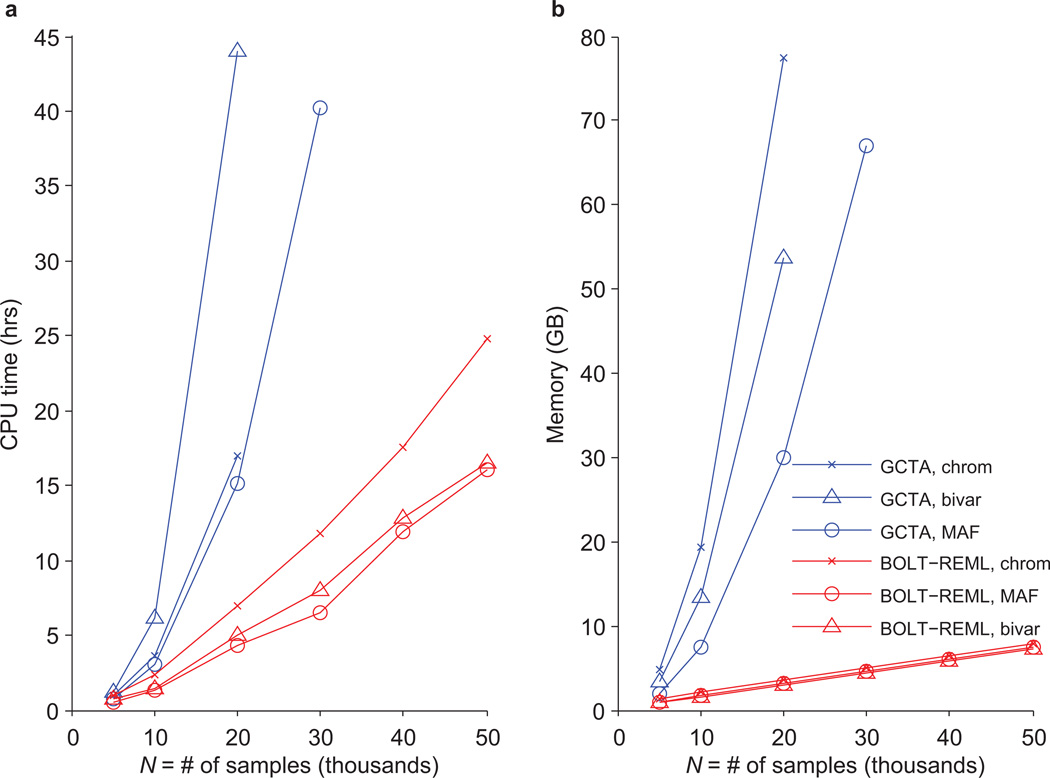

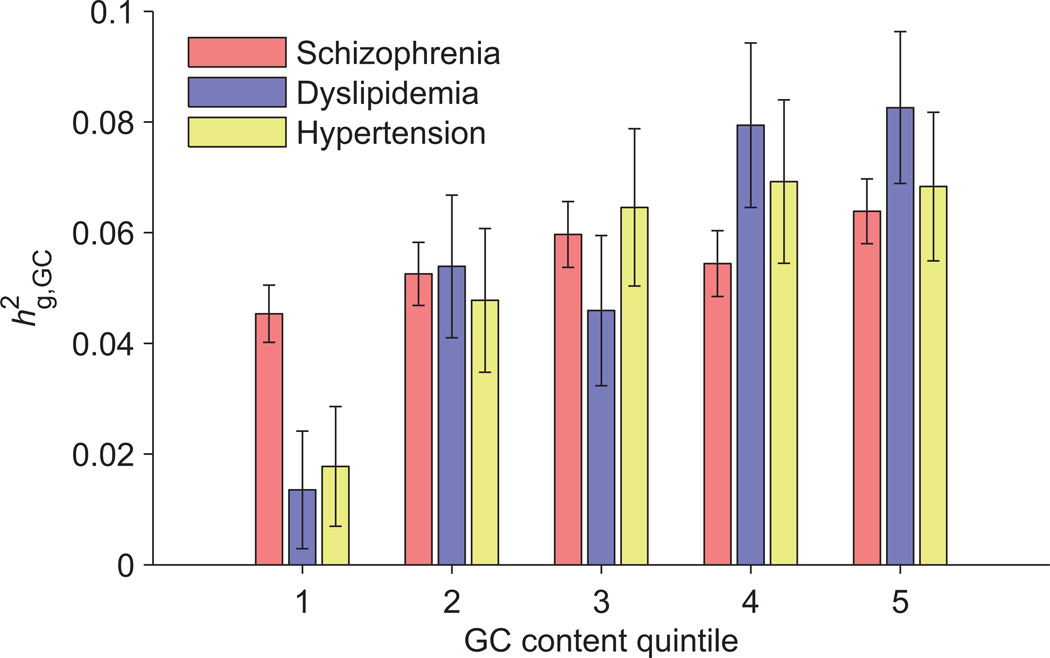

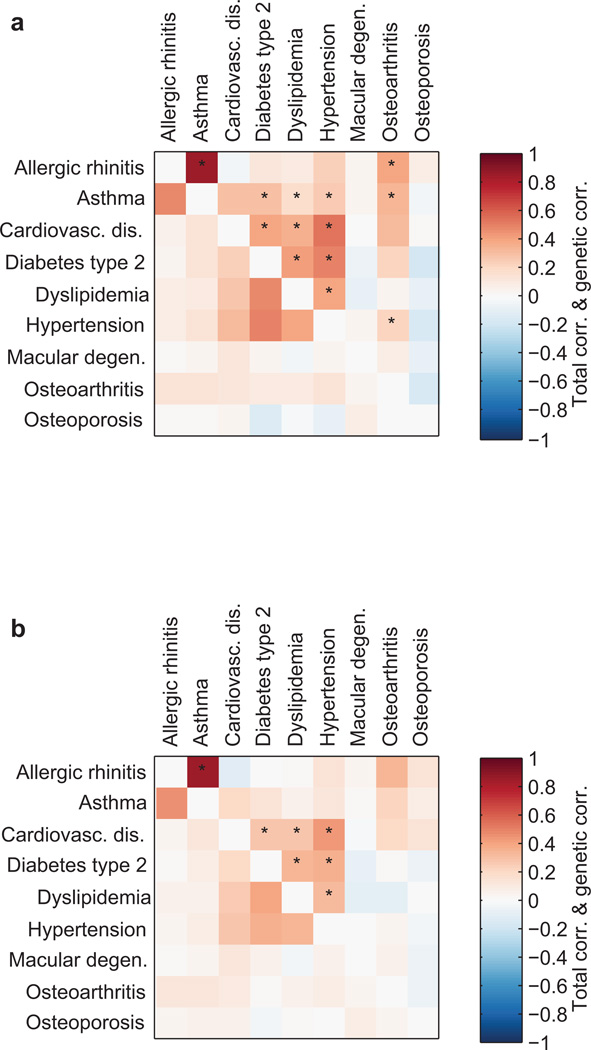
References
References (Online Methods)
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
