

An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development
- PMID: 26523971
- PMCID: PMC4955611
- DOI: 10.1038/nm.3982
An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development
Abstract
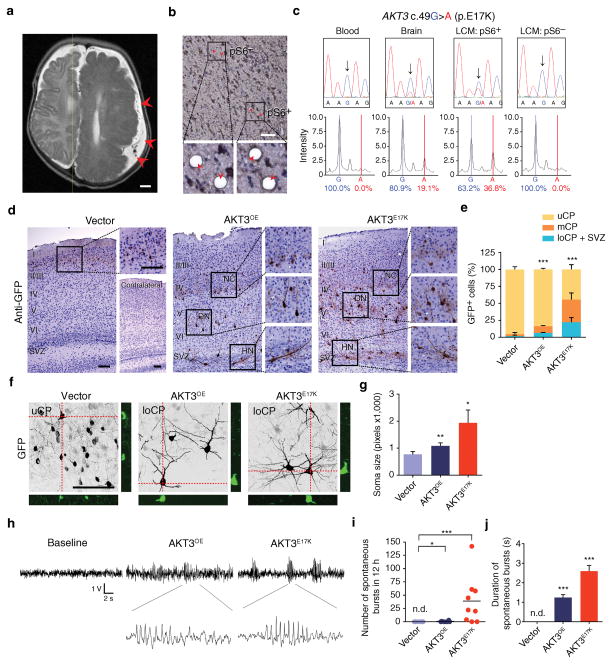
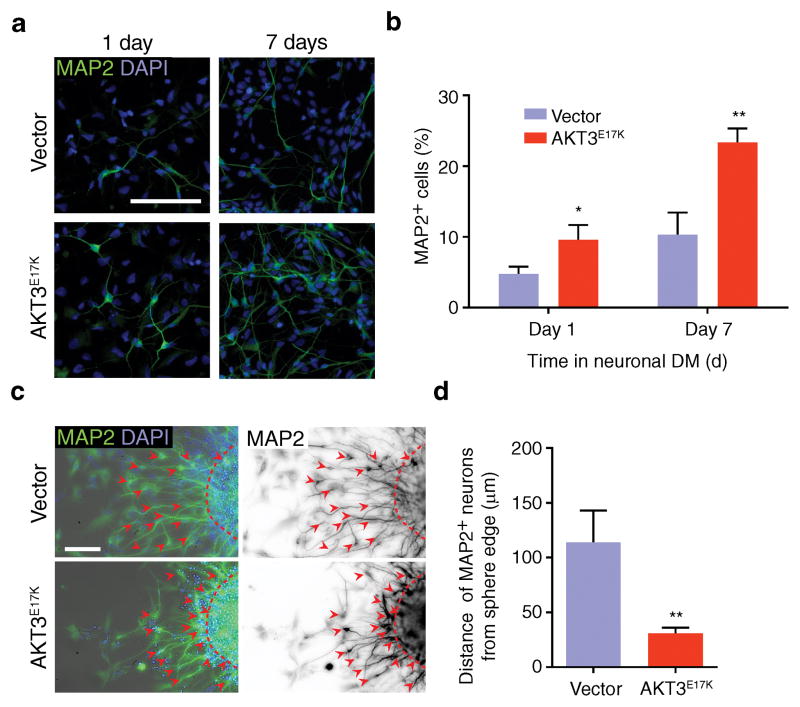
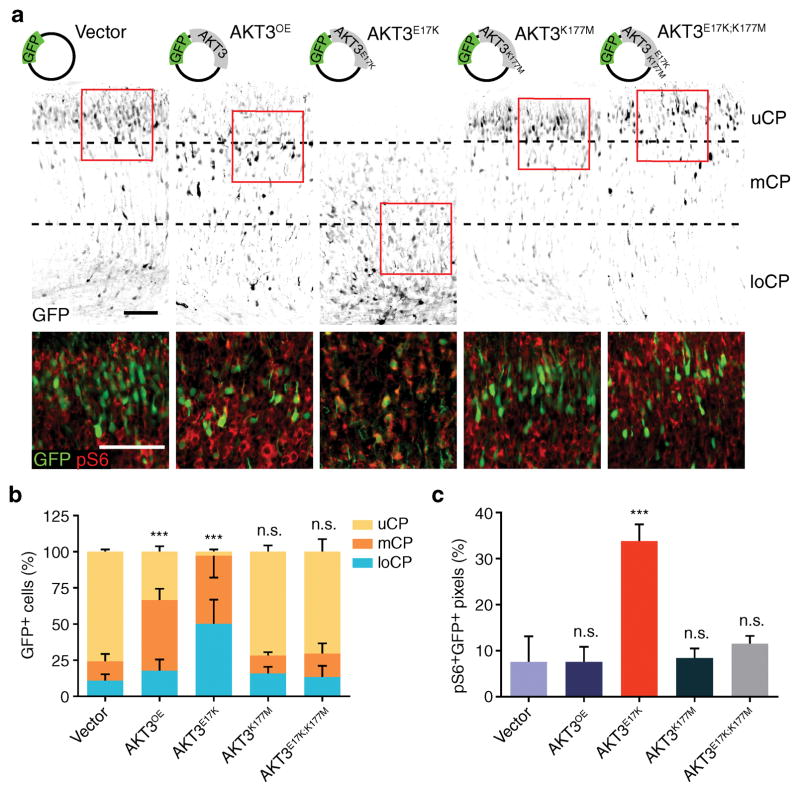
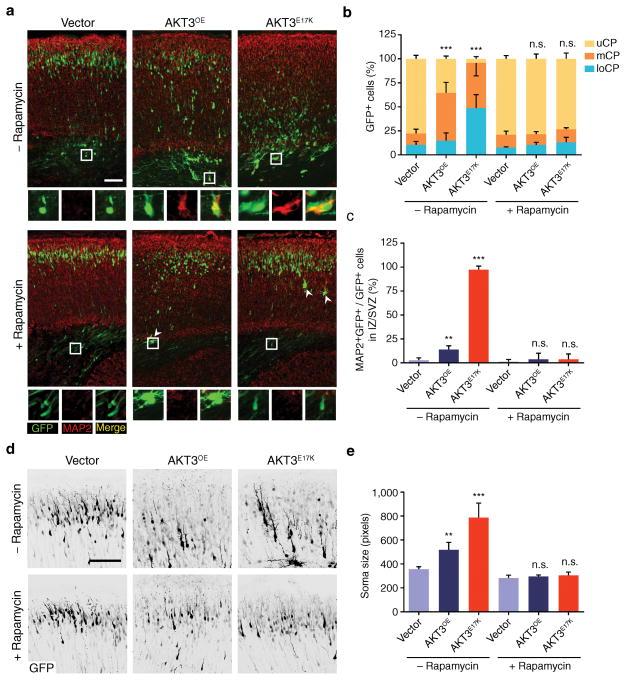
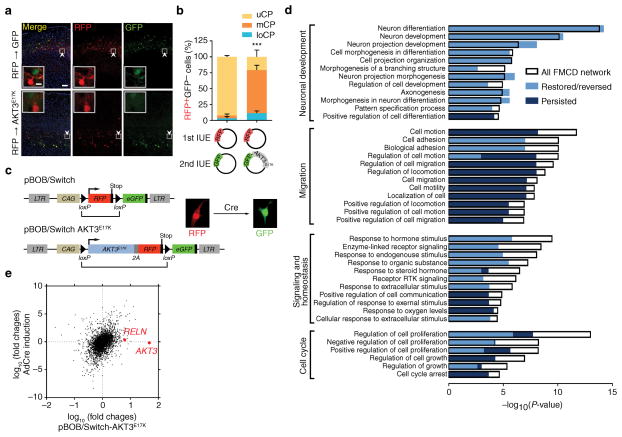
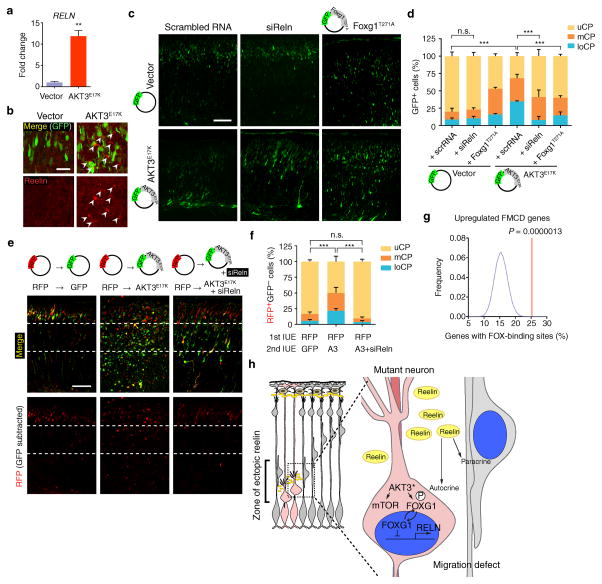
Focal malformations of cortical development (FMCDs) account for the majority of drug-resistant pediatric epilepsy. Postzygotic somatic mutations activating the phosphatidylinositol-4,5-bisphosphate-3-kinase (PI3K)-protein kinase B (AKT)-mammalian target of rapamycin (mTOR) pathway are found in a wide range of brain diseases, including FMCDs. It remains unclear how a mutation in a small fraction of cells disrupts the architecture of the entire hemisphere. Within human FMCD-affected brain, we found that cells showing activation of the PI3K-AKT-mTOR pathway were enriched for the AKT3(E17K) mutation. Introducing the FMCD-causing mutation into mouse brain resulted in electrographic seizures and impaired hemispheric architecture. Mutation-expressing neural progenitors showed misexpression of reelin, which led to a non-cell autonomous migration defect in neighboring cells, due at least in part to derepression of reelin transcription in a manner dependent on the forkhead box (FOX) transcription factor FOXG1. Treatments aimed at either blocking downstream AKT signaling or inactivating reelin restored migration. These findings suggest a central AKT-FOXG1-reelin signaling pathway in FMCD and support pathway inhibitors as potential treatments or therapies for some forms of focal epilepsy.
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
