

Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production
- PMID: 26524591
- PMCID: PMC4639987
- DOI: 10.1172/JCI81260
Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production
Erratum in
-
Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production.J Clin Invest. 2016 Feb;126(2):795. doi: 10.1172/JCI86020. Epub 2016 Feb 1. J Clin Invest. 2016. PMID: 26829627 Free PMC article. No abstract available.
Abstract
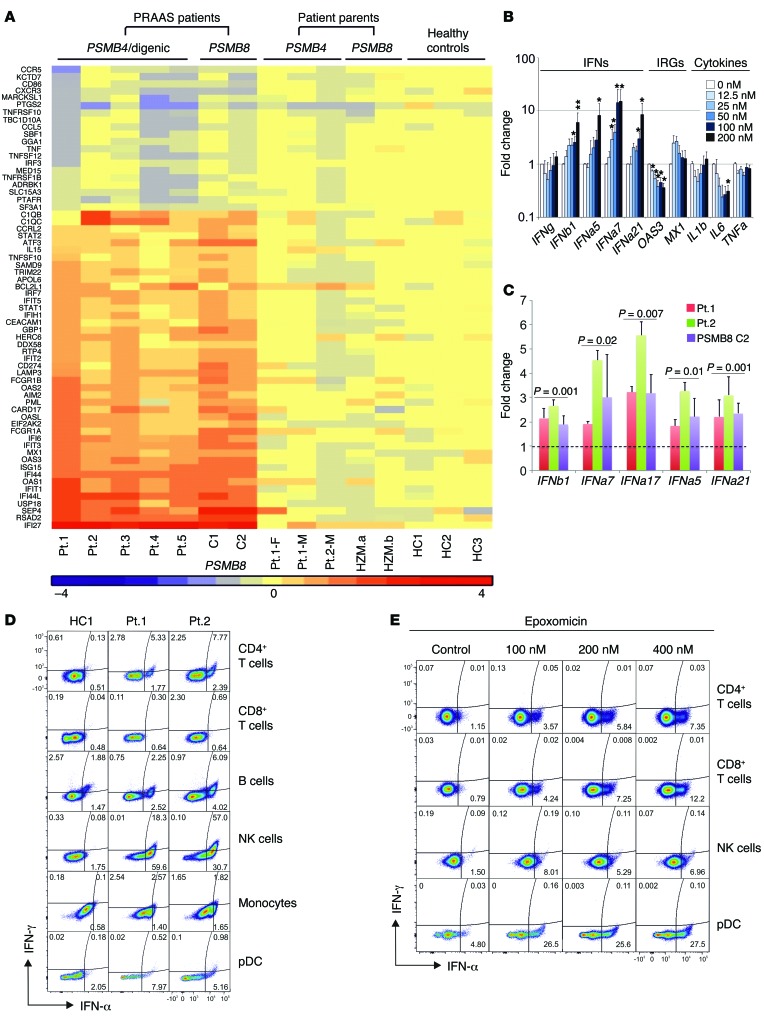
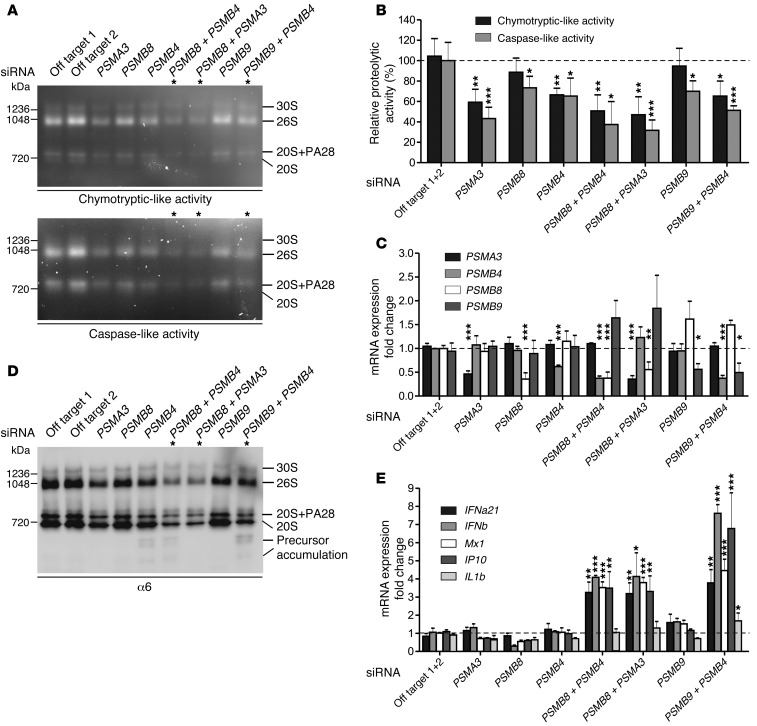
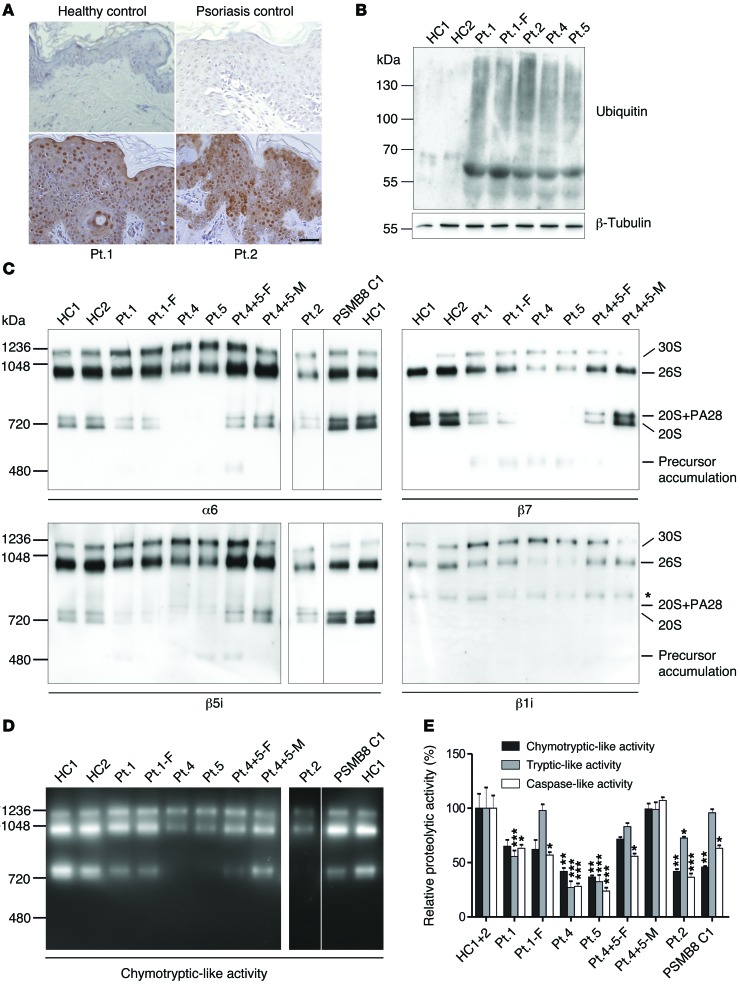
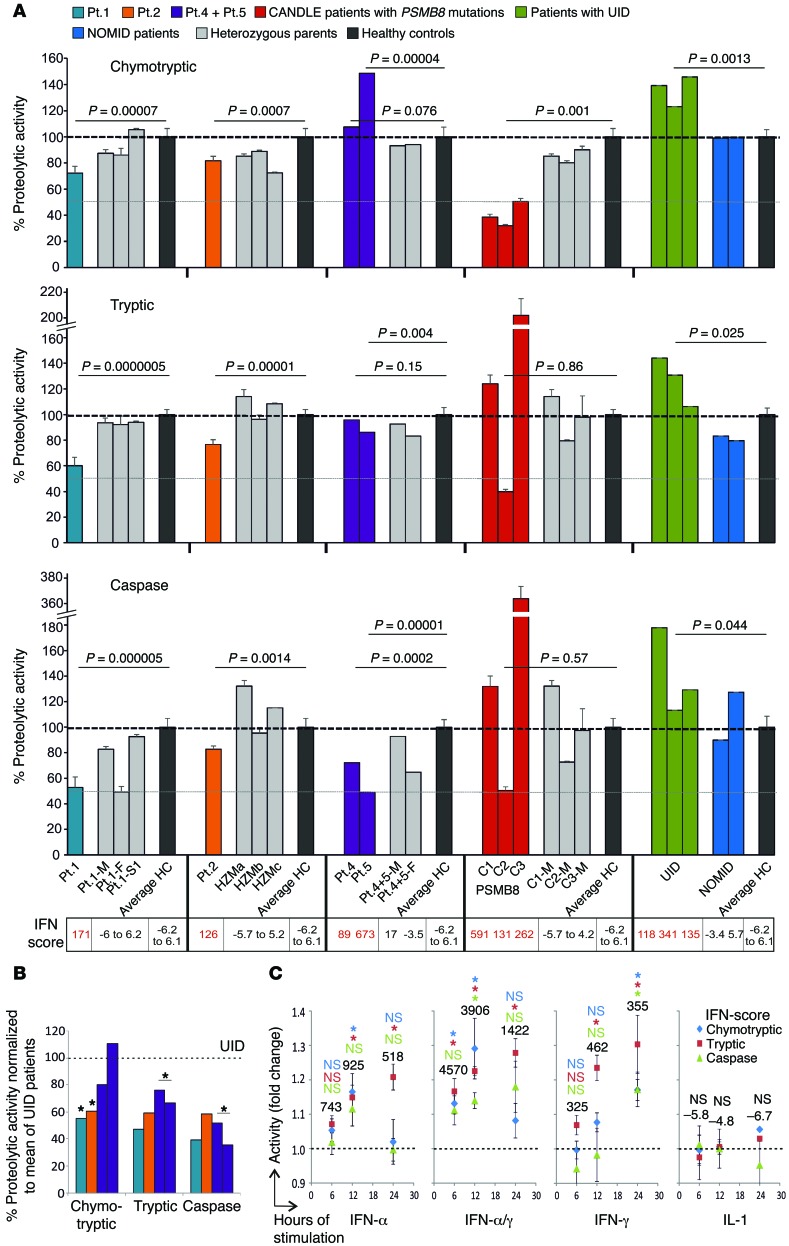
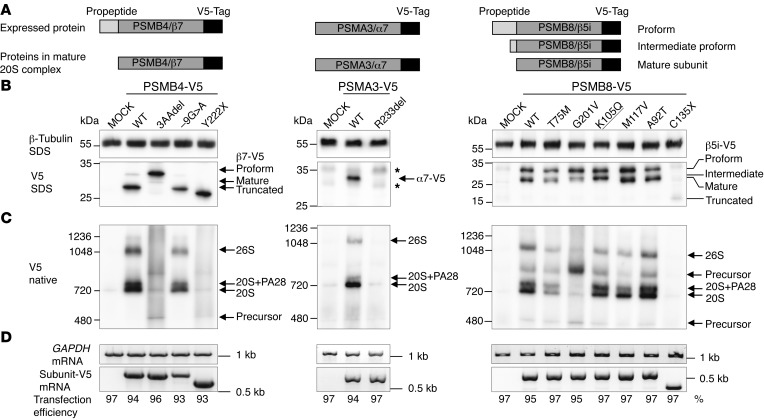
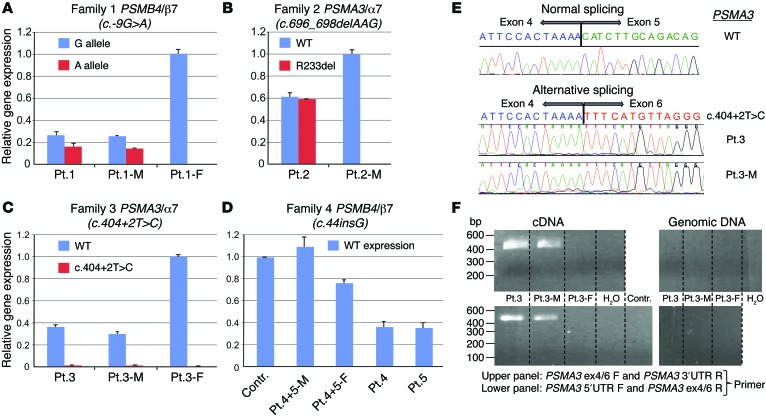
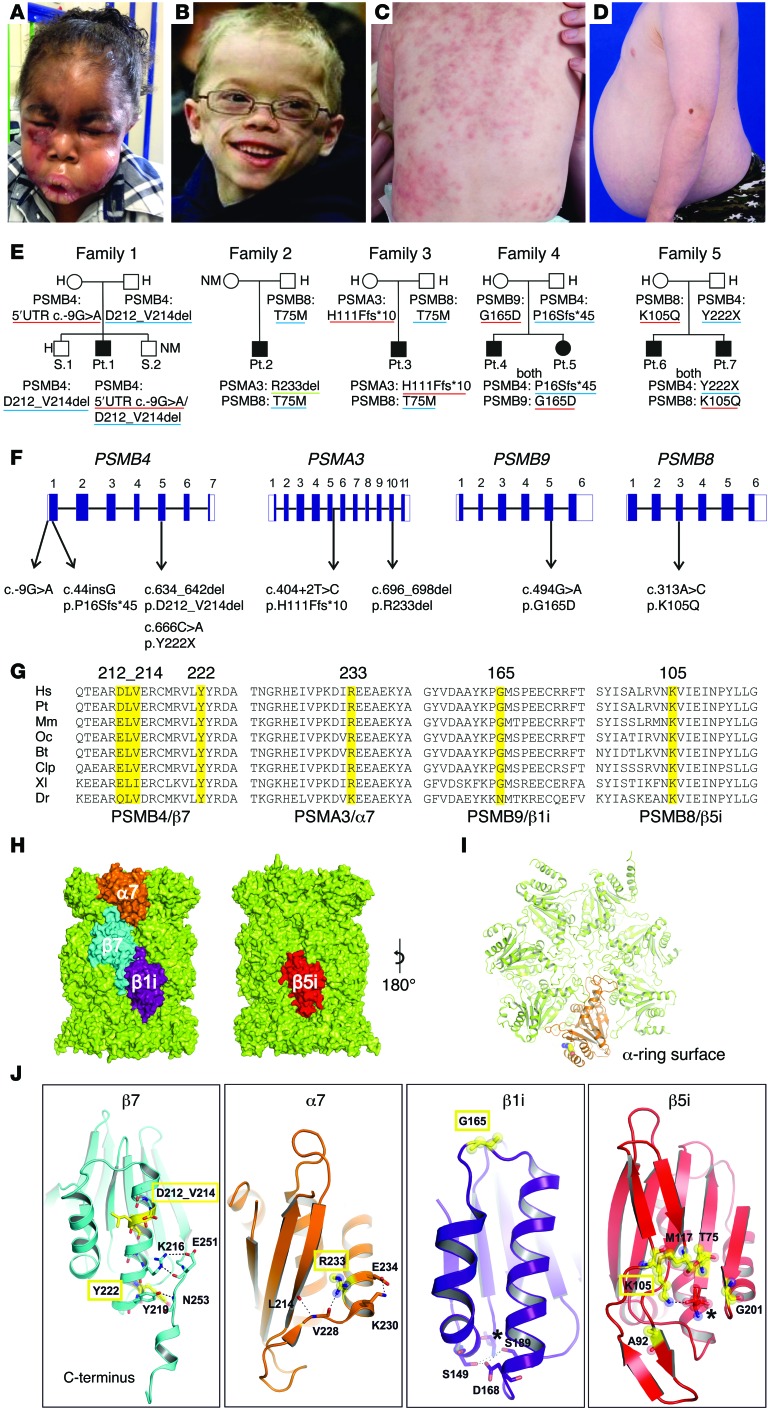
Autosomal recessive mutations in proteasome subunit β 8 (PSMB8), which encodes the inducible proteasome subunit β5i, cause the immune-dysregulatory disease chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE), which is classified as a proteasome-associated autoinflammatory syndrome (PRAAS). Here, we identified 8 mutations in 4 proteasome genes, PSMA3 (encodes α7), PSMB4 (encodes β7), PSMB9 (encodes β1i), and proteasome maturation protein (POMP), that have not been previously associated with disease and 1 mutation in PSMB8 that has not been previously reported. One patient was compound heterozygous for PSMB4 mutations, 6 patients from 4 families were heterozygous for a missense mutation in 1 inducible proteasome subunit and a mutation in a constitutive proteasome subunit, and 1 patient was heterozygous for a POMP mutation, thus establishing a digenic and autosomal dominant inheritance pattern of PRAAS. Function evaluation revealed that these mutations variably affect transcription, protein expression, protein folding, proteasome assembly, and, ultimately, proteasome activity. Moreover, defects in proteasome formation and function were recapitulated by siRNA-mediated knockdown of the respective subunits in primary fibroblasts from healthy individuals. Patient-isolated hematopoietic and nonhematopoietic cells exhibited a strong IFN gene-expression signature, irrespective of genotype. Additionally, chemical proteasome inhibition or progressive depletion of proteasome subunit gene transcription with siRNA induced transcription of type I IFN genes in healthy control cells. Our results provide further insight into CANDLE genetics and link global proteasome dysfunction to increased type I IFN production.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
