

The role of macropinocytosis in the propagation of protein aggregation associated with neurodegenerative diseases
- PMID: 26528186
- PMCID: PMC4607857
- DOI: 10.3389/fphys.2015.00277
The role of macropinocytosis in the propagation of protein aggregation associated with neurodegenerative diseases
Abstract
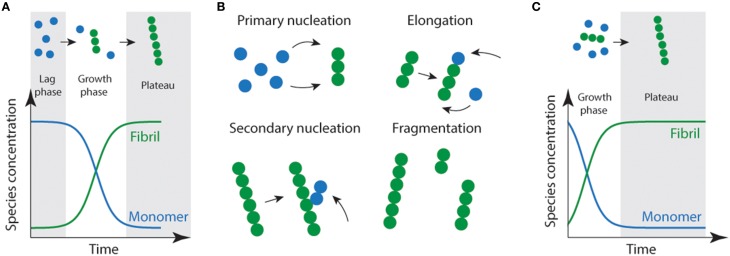
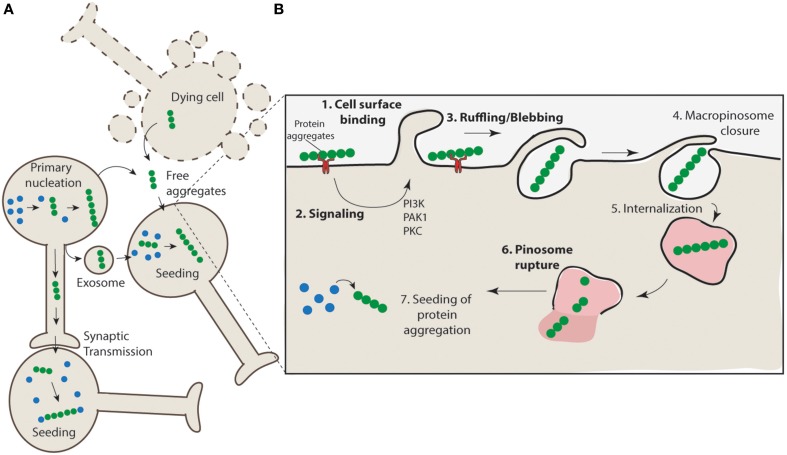
With the onset of the rapidly aging population, the impact of age related neurodegenerative diseases is becoming a predominant health and economic concern. Neurodegenerative diseases such as Alzheimer's disease, Creutzfeldt-Jakob disease (CJD), Parkinson's disease, Huntington's disease, frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS) result from the loss of a specific subsets of neurons, which is closely associated with accumulation and deposition of specific protein aggregates. Protein aggregation, or fibril formation, is a well-studied phenomenon that occurs in a nucleation-dependent growth reaction. Recently, there has been a swell of literature implicating protein aggregation and its ability to propagate cell-to-cell in the rapid progression of these diseases. In order for protein aggregation to be kindled in recipient cells it is a requisite that aggregates must be able to be released from one cell and then taken up by others. In this article we will explore the relationship between protein aggregates, their propagation and the role of macropinocytosis in their uptake. We highlight the ability of neurons to undergo stimulated macropinocytosis and identify potential therapeutic targets.
Keywords: amyloid fibrils; endocytosis; macropinocytosis; propagation; protein aggregation.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous