

Inactivating the permanent neonatal diabetes gene Mnx1 switches insulin-producing β-cells to a δ-like fate and reveals a facultative proliferative capacity in aged β-cells
- PMID: 26534984
- PMCID: PMC4647212
- DOI: 10.1242/dev.126011
Inactivating the permanent neonatal diabetes gene Mnx1 switches insulin-producing β-cells to a δ-like fate and reveals a facultative proliferative capacity in aged β-cells
Abstract
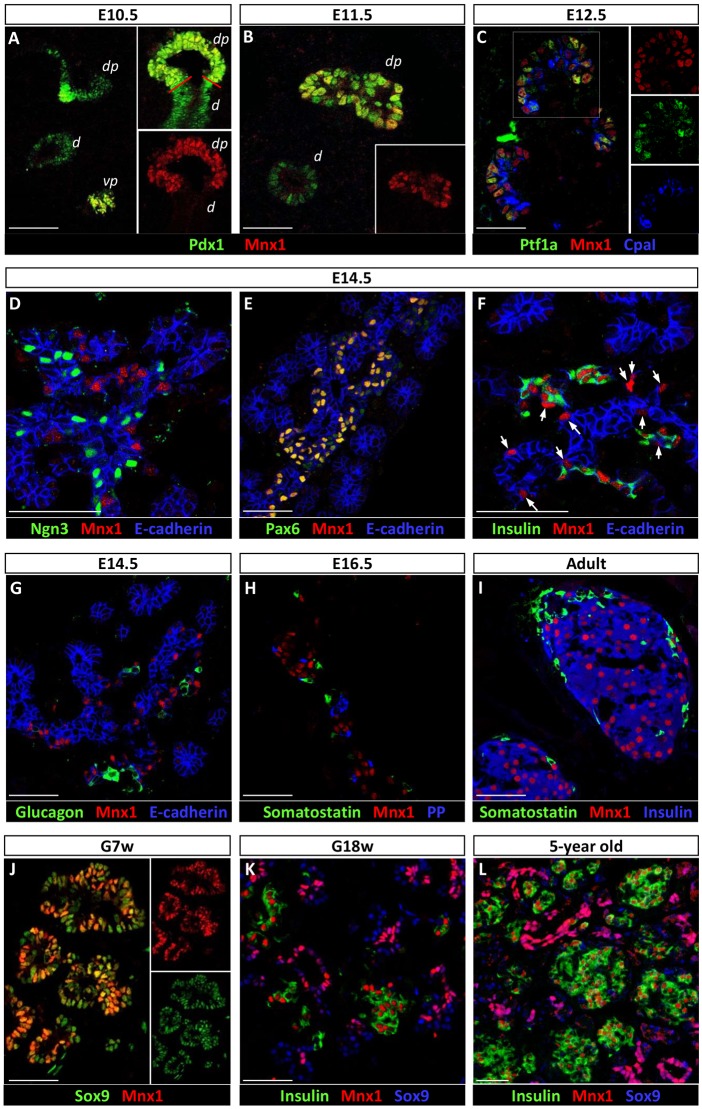
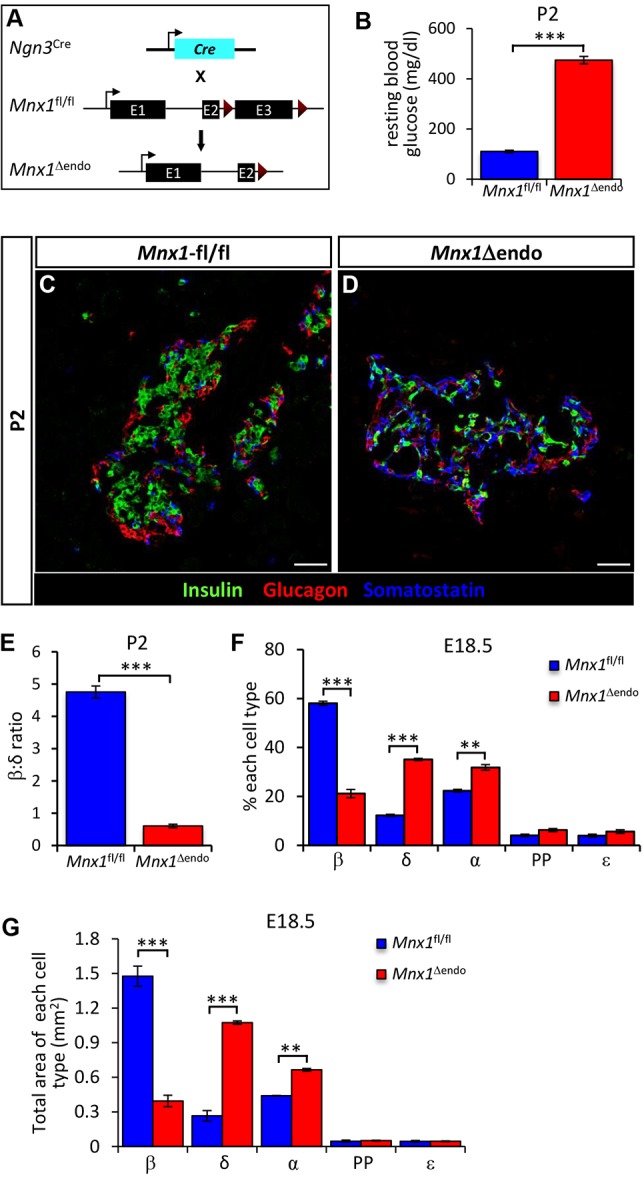
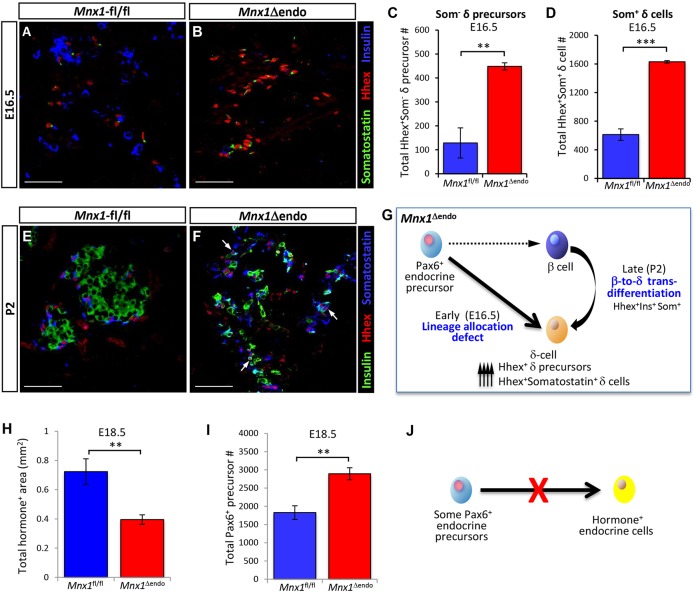
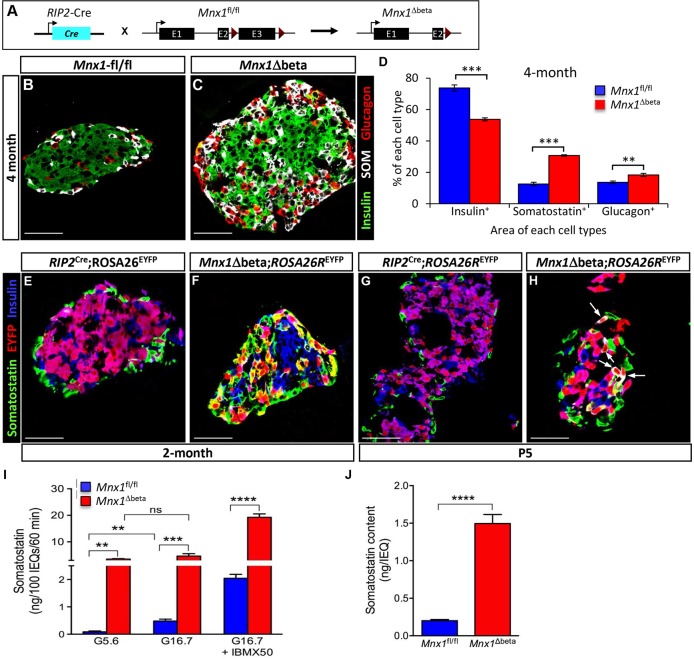
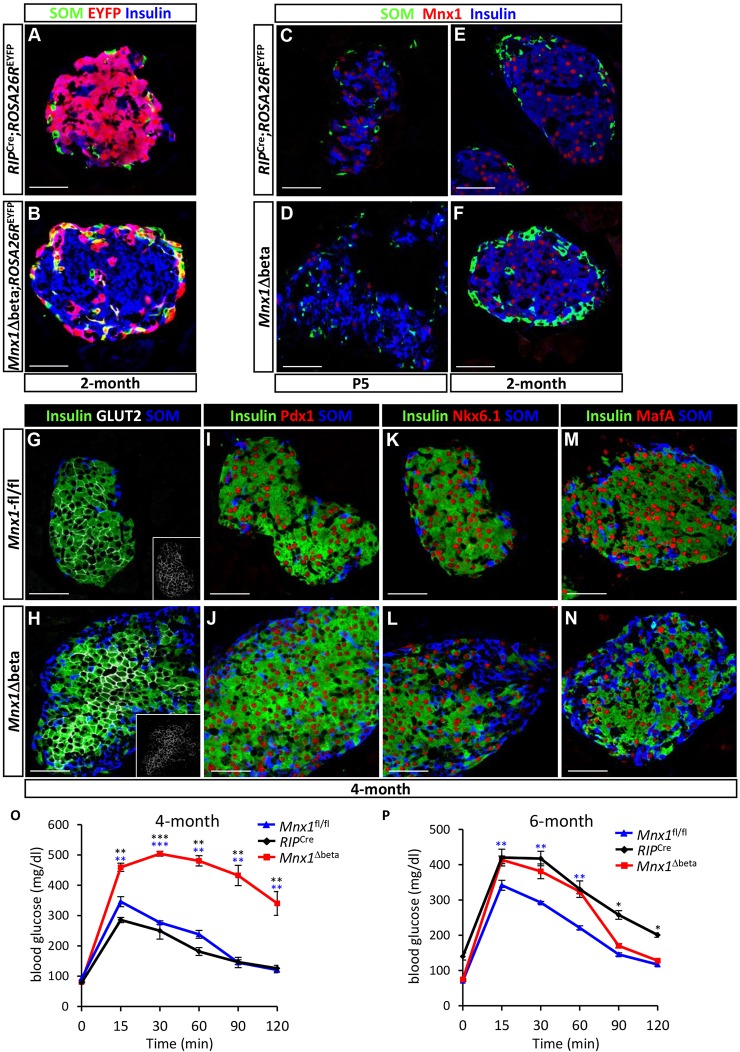
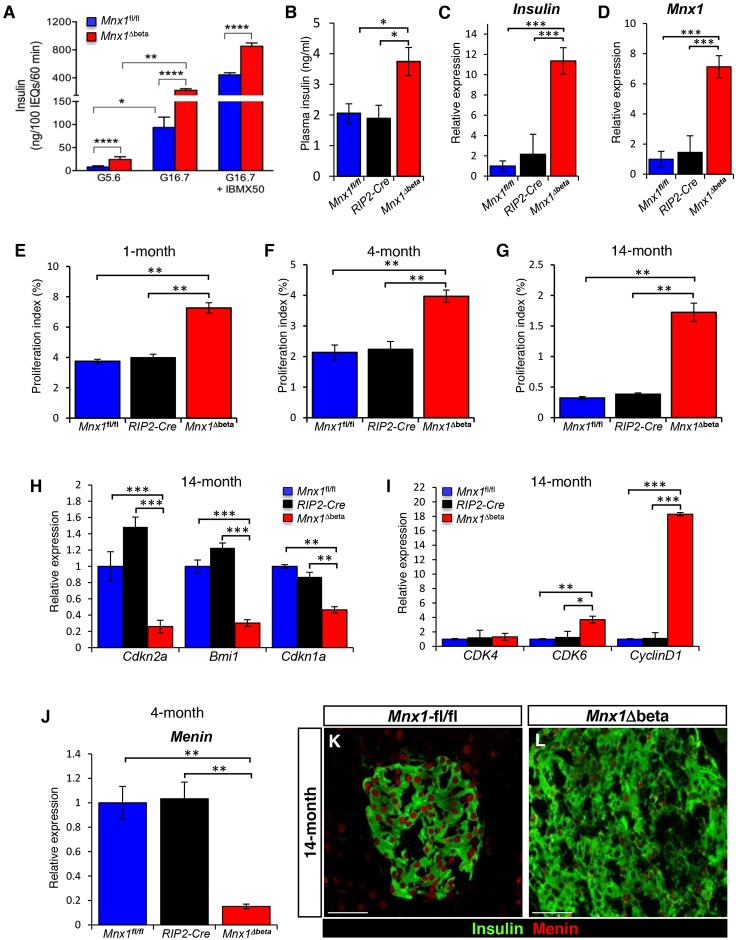
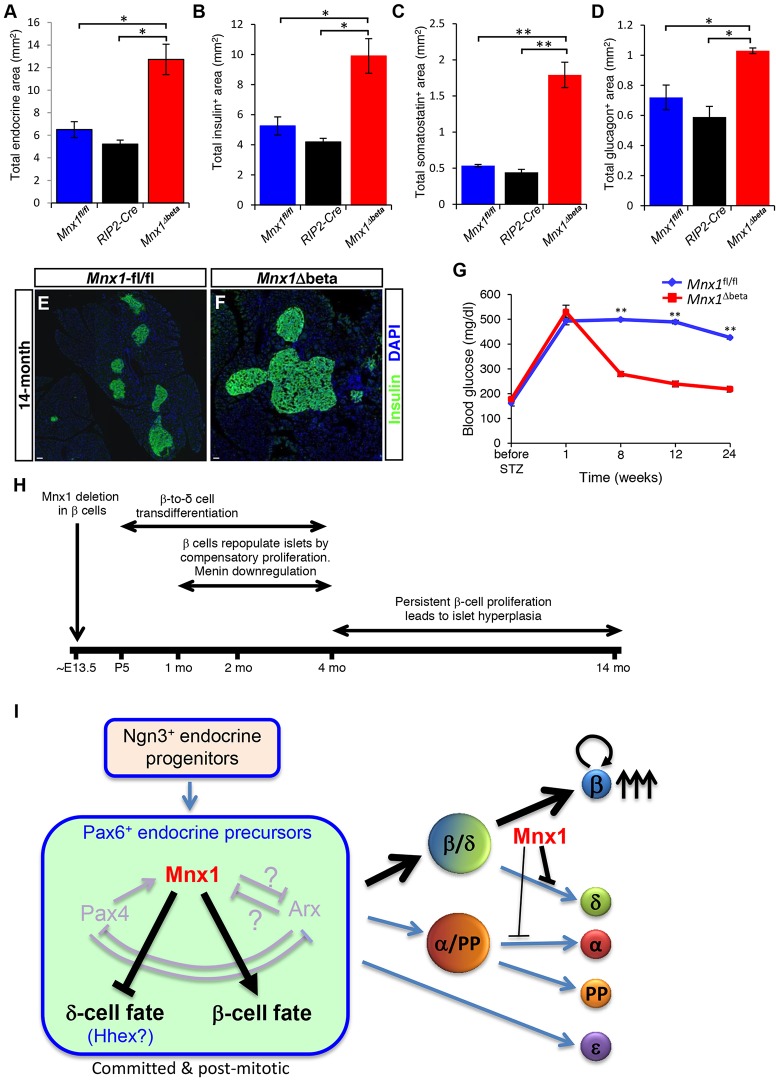
Homozygous Mnx1 mutation causes permanent neonatal diabetes in humans, but via unknown mechanisms. Our systematic and longitudinal analysis of Mnx1 function during murine pancreas organogenesis and into the adult uncovered novel stage-specific roles for Mnx1 in endocrine lineage allocation and β-cell fate maintenance. Inactivation in the endocrine-progenitor stage shows that Mnx1 promotes β-cell while suppressing δ-cell differentiation programs, and is crucial for postnatal β-cell fate maintenance. Inactivating Mnx1 in embryonic β-cells (Mnx1(Δbeta)) caused β-to-δ-like cell transdifferentiation, which was delayed until postnatal stages. In the latter context, β-cells escaping Mnx1 inactivation unexpectedly upregulated Mnx1 expression and underwent an age-independent persistent proliferation. Escaper β-cells restored, but then eventually surpassed, the normal pancreatic β-cell mass, leading to islet hyperplasia in aged mice. In vitro analysis of islets isolated from Mnx1(Δbeta) mice showed higher insulin secretory activity and greater insulin mRNA content than in wild-type islets. Mnx1(Δbeta) mice also showed a much faster return to euglycemia after β-cell ablation, suggesting that the new β-cells derived from the escaper population are functional. Our findings identify Mnx1 as an important factor in β-cell differentiation and proliferation, with the potential for targeting to increase the number of endogenous β-cells for diabetes therapy.
Keywords: Endocrine lineage diversification; Islet; β-cell fate maintenance; β-cell proliferation; β-cell versus δ-cell fate selection.
© 2015. Published by The Company of Biologists Ltd.
Conflict of interest statement
The authors declare no competing or financial interests.
Figures
References
-
- Bonnefond A., Vaillant E., Philippe J., Skrobek B., Lobbens S., Yengo L., Huyvaert M., Cavé H., Busiah K., Scharfmann R. et al. (2013). Transcription factor gene MNX1 is a novel cause of permanent neonatal diabetes in a consanguineous family. Diabetes Metab. 39, 276-280. 10.1016/j.diabet.2013.02.007 - DOI - PubMed
-
- Brissova M., Aamodt K., Brahmachary P., Prasad N., Hong J.-Y., Dai C., Mellati M., Shostak A., Poffenberger G., Aramandla R. et al. (2014). Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes β cell regeneration. Cell Metab. 19, 498-511. 10.1016/j.cmet.2014.02.001 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- P60 DK020593/DK/NIDDK NIH HHS/United States
- U19 DK 042502/DK/NIDDK NIH HHS/United States
- U19 DK042502/DK/NIDDK NIH HHS/United States
- UC4 DK104211/DK/NIDDK NIH HHS/United States
- DK104211/DK/NIDDK NIH HHS/United States
- DK58404/DK/NIDDK NIH HHS/United States
- P30 DK058404/DK/NIDDK NIH HHS/United States
- U01 DK089570/DK/NIDDK NIH HHS/United States
- U01 DK072473/DK/NIDDK NIH HHS/United States
- U01 DK089572/DK/NIDDK NIH HHS/United States
- U01 DK 089570/DK/NIDDK NIH HHS/United States
- DK59637/DK/NIDDK NIH HHS/United States
- P30 DK020593/DK/NIDDK NIH HHS/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- DK72473/DK/NIDDK NIH HHS/United States
- DK89572/DK/NIDDK NIH HHS/United States
- CA68485/CA/NCI NIH HHS/United States
- DK20593/DK/NIDDK NIH HHS/United States
- U24 DK059637/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
