

ADCY5-related dyskinesia: Broader spectrum and genotype-phenotype correlations
- PMID: 26537056
- PMCID: PMC4676753
- DOI: 10.1212/WNL.0000000000002058
ADCY5-related dyskinesia: Broader spectrum and genotype-phenotype correlations
Abstract
Objective: To investigate the clinical spectrum and distinguishing features of adenylate cyclase 5 (ADCY5)-related dyskinesia and genotype-phenotype relationship.
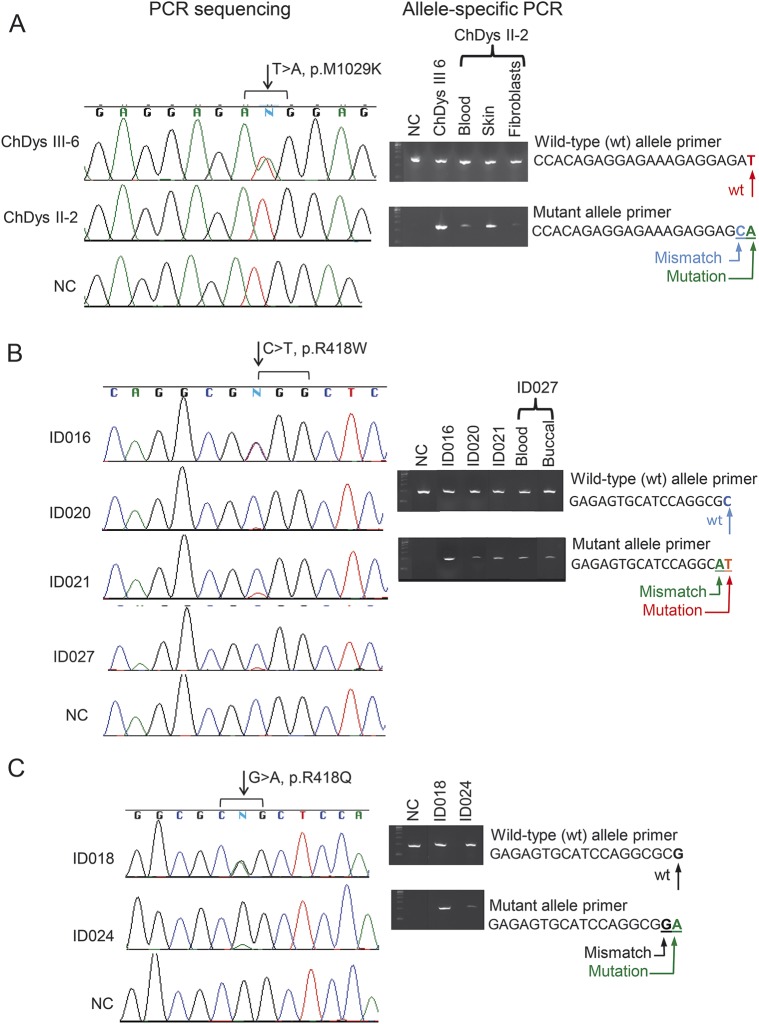
Methods: We analyzed ADCY5 in patients with choreiform or dystonic movements by exome or targeted sequencing. Suspected mosaicism was confirmed by allele-specific amplification. We evaluated clinical features in our 50 new and previously reported cases.
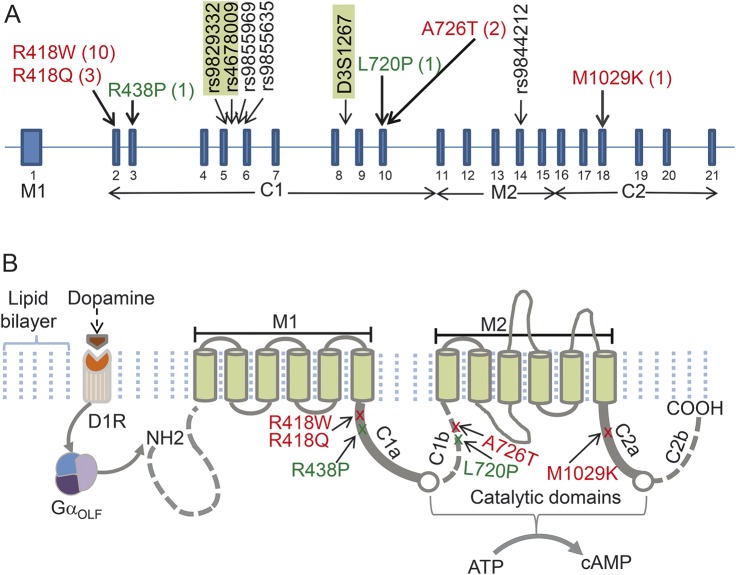
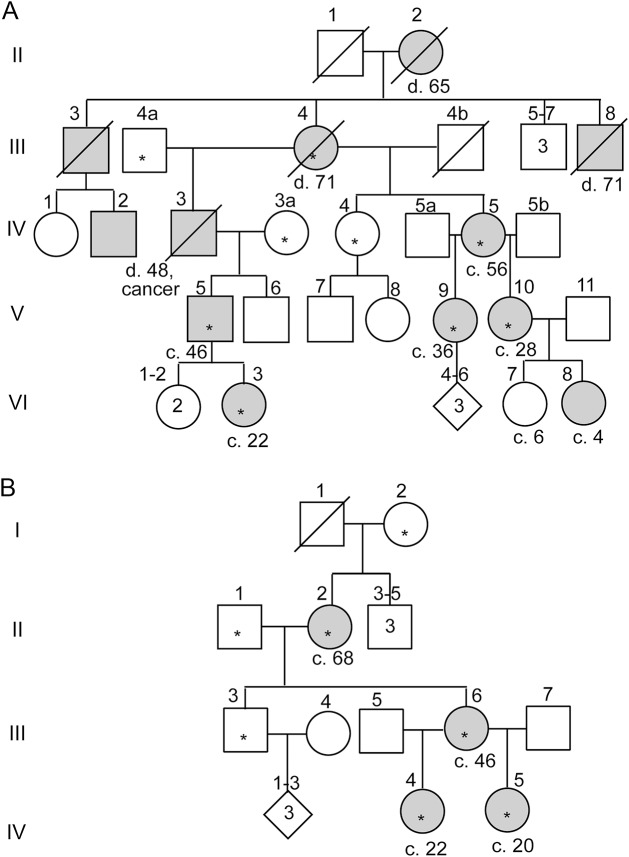
Results: We identified 3 new families and 12 new sporadic cases with ADCY5 mutations. These mutations cause a mixed hyperkinetic disorder that includes dystonia, chorea, and myoclonus, often with facial involvement. The movements are sometimes painful and show episodic worsening on a fluctuating background. Many patients have axial hypotonia. In 2 unrelated families, a p.A726T mutation in the first cytoplasmic domain (C1) causes a relatively mild disorder of prominent facial and hand dystonia and chorea. Mutations p.R418W or p.R418Q in C1, de novo in 13 individuals and inherited in 1, produce a moderate to severe disorder with axial hypotonia, limb hypertonia, paroxysmal nocturnal or diurnal dyskinesia, chorea, myoclonus, and intermittent facial dyskinesia. Somatic mosaicism is usually associated with a less severe phenotype. In one family, a p.M1029K mutation in the C2 domain causes severe dystonia, hypotonia, and chorea. The progenitor, whose childhood-onset episodic movement disorder almost disappeared in adulthood, was mosaic for the mutation.
Conclusions: ADCY5-related dyskinesia is a childhood-onset disorder with a wide range of hyperkinetic abnormal movements. Genotype-specific correlations and mosaicism play important roles in the phenotypic variability. Recurrent mutations suggest particular functional importance of residues 418 and 726 in disease pathogenesis.
© 2015 American Academy of Neurology.
Figures
References
-
- Hanoune J, Pouille Y, Tzavara E, et al. Adenylyl cyclases: structure, regulation and function in an enzyme superfamily. Mol Cell Endocrinol 1997;128:179–194. - PubMed
-
- Iwamoto T, Okumura S, Iwatsubo K, et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J Biol Chem 2003;278:16936–16940. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous