

Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease
- PMID: 26539891
- PMCID: PMC4824012
- DOI: 10.1016/j.neuron.2015.09.048
Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease
Abstract
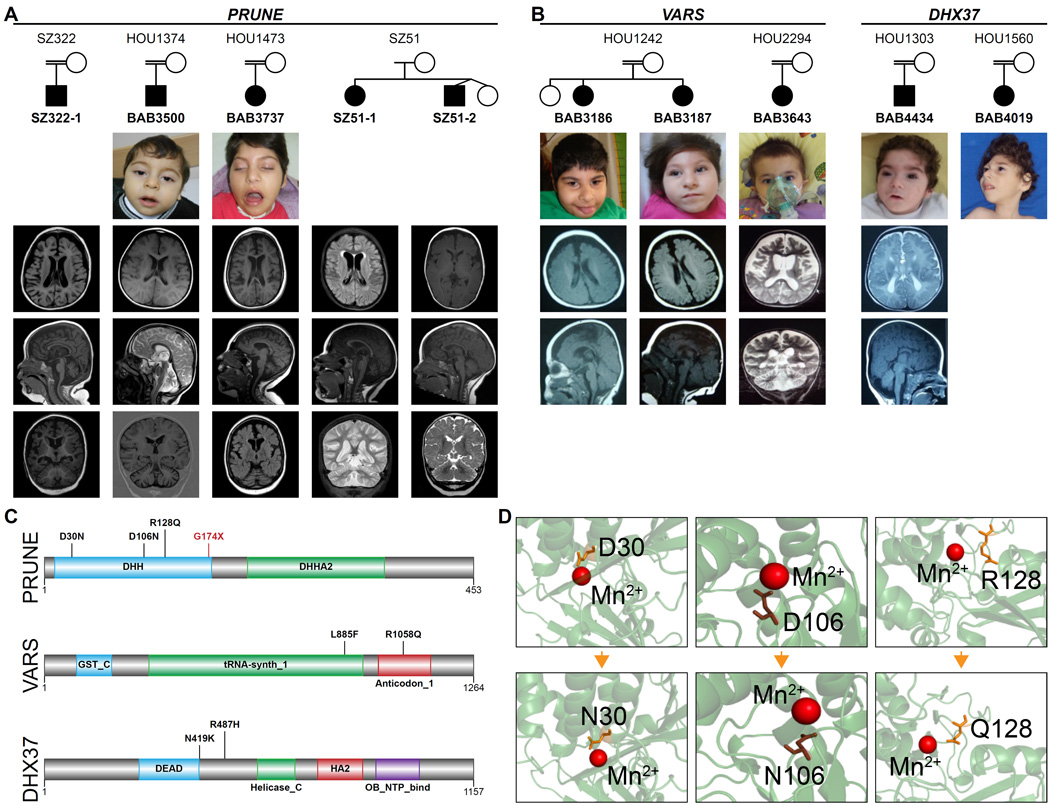
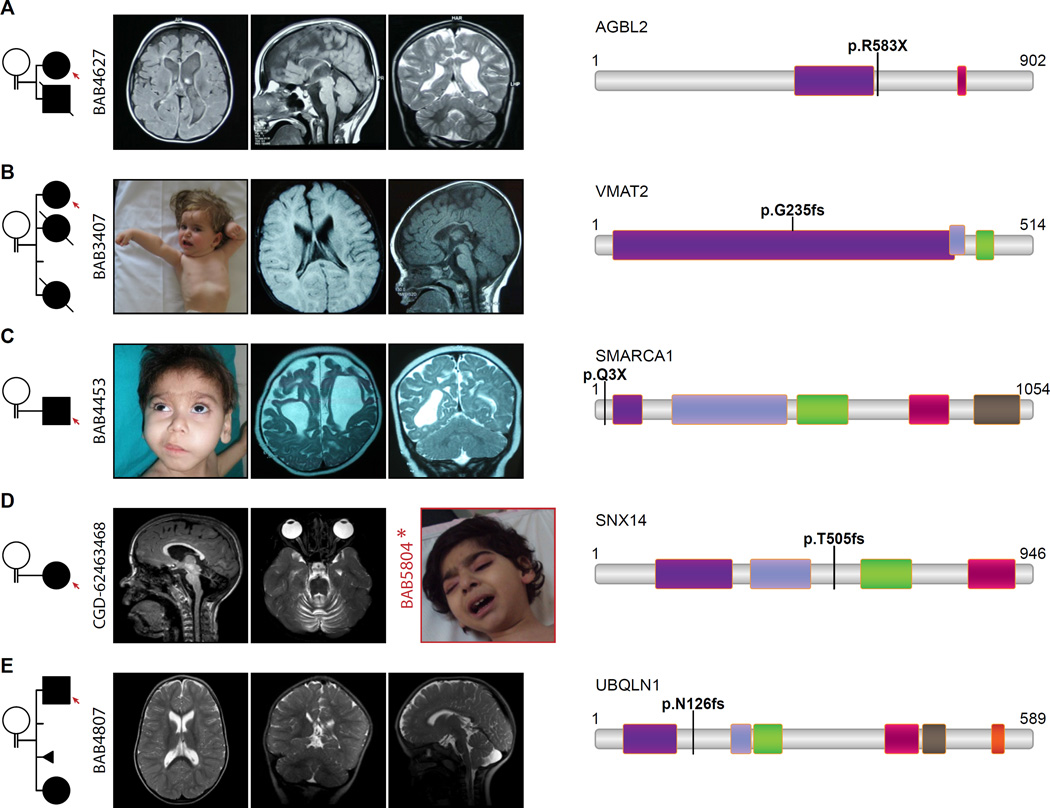
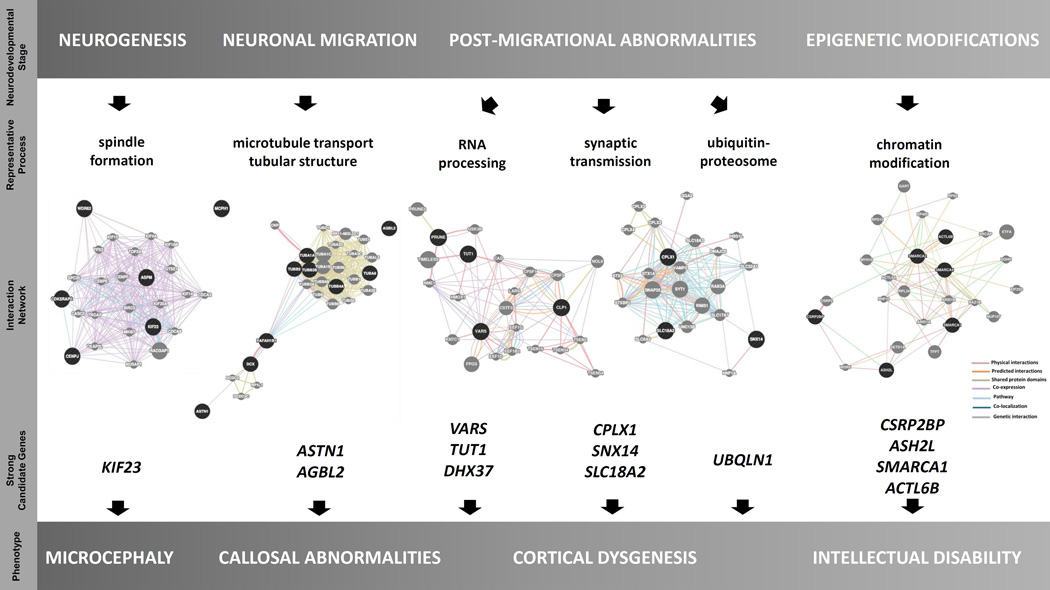
Development of the human nervous system involves complex interactions among fundamental cellular processes and requires a multitude of genes, many of which remain to be associated with human disease. We applied whole exome sequencing to 128 mostly consanguineous families with neurogenetic disorders that often included brain malformations. Rare variant analyses for both single nucleotide variant (SNV) and copy number variant (CNV) alleles allowed for identification of 45 novel variants in 43 known disease genes, 41 candidate genes, and CNVs in 10 families, with an overall potential molecular cause identified in >85% of families studied. Among the candidate genes identified, we found PRUNE, VARS, and DHX37 in multiple families and homozygous loss-of-function variants in AGBL2, SLC18A2, SMARCA1, UBQLN1, and CPLX1. Neuroimaging and in silico analysis of functional and expression proximity between candidate and known disease genes allowed for further understanding of genetic networks underlying specific types of brain malformations.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
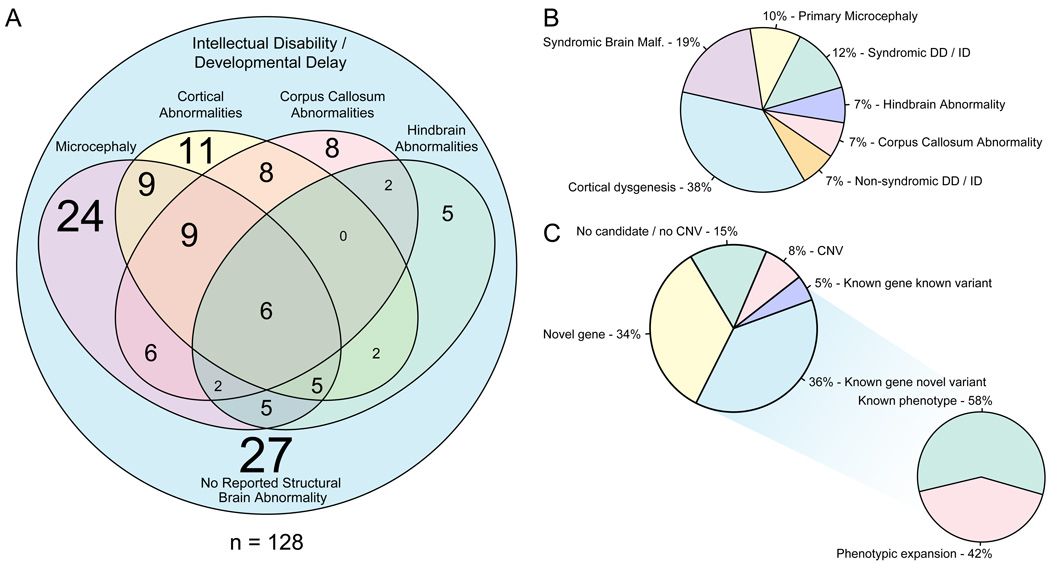
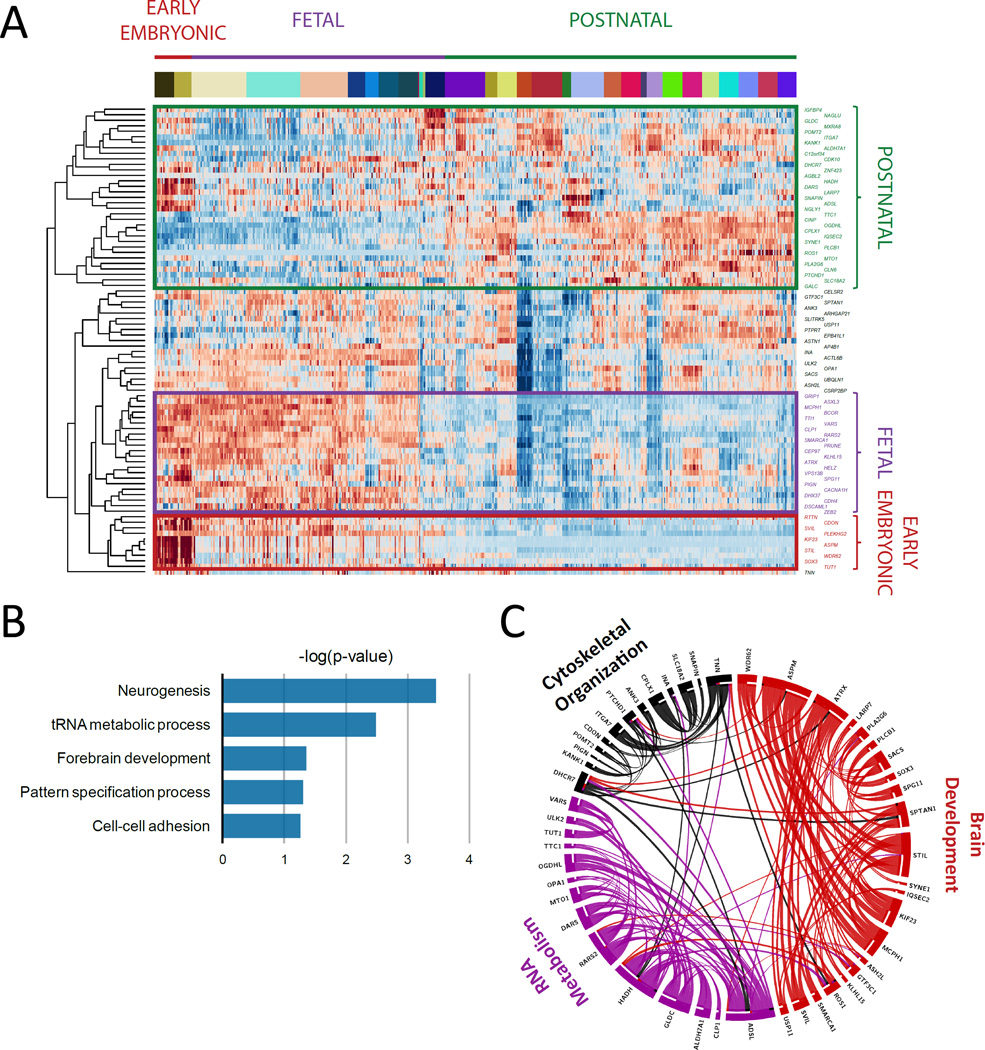
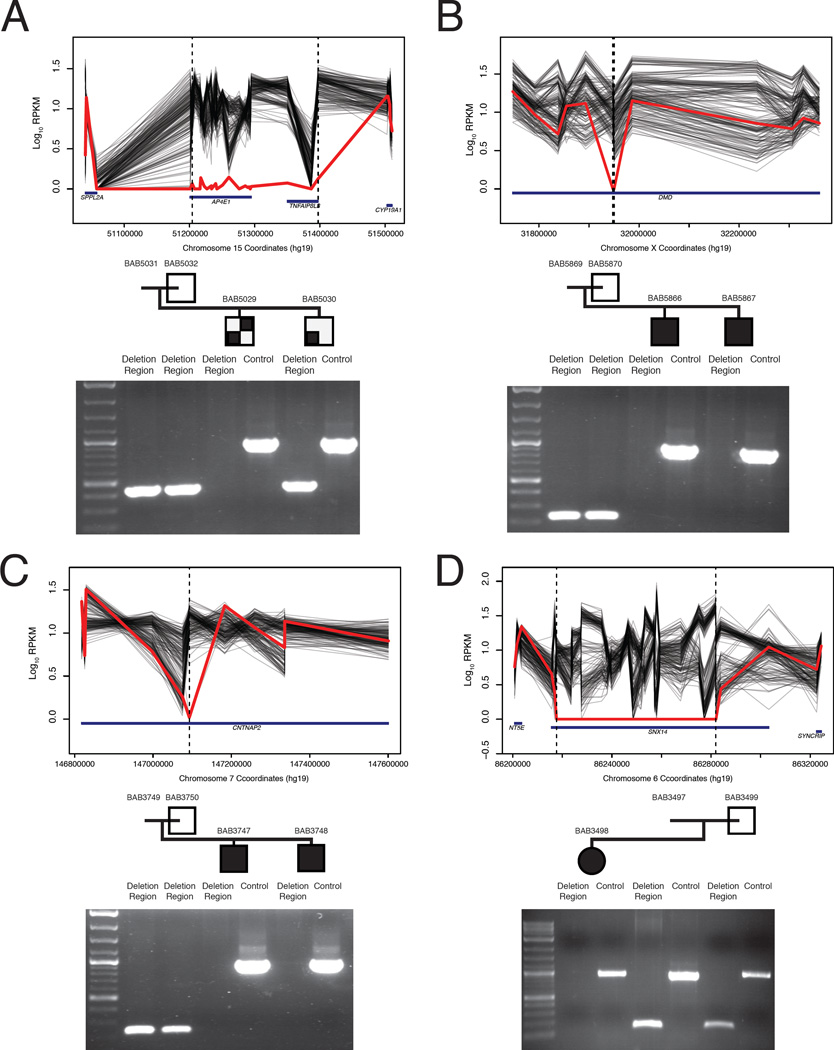
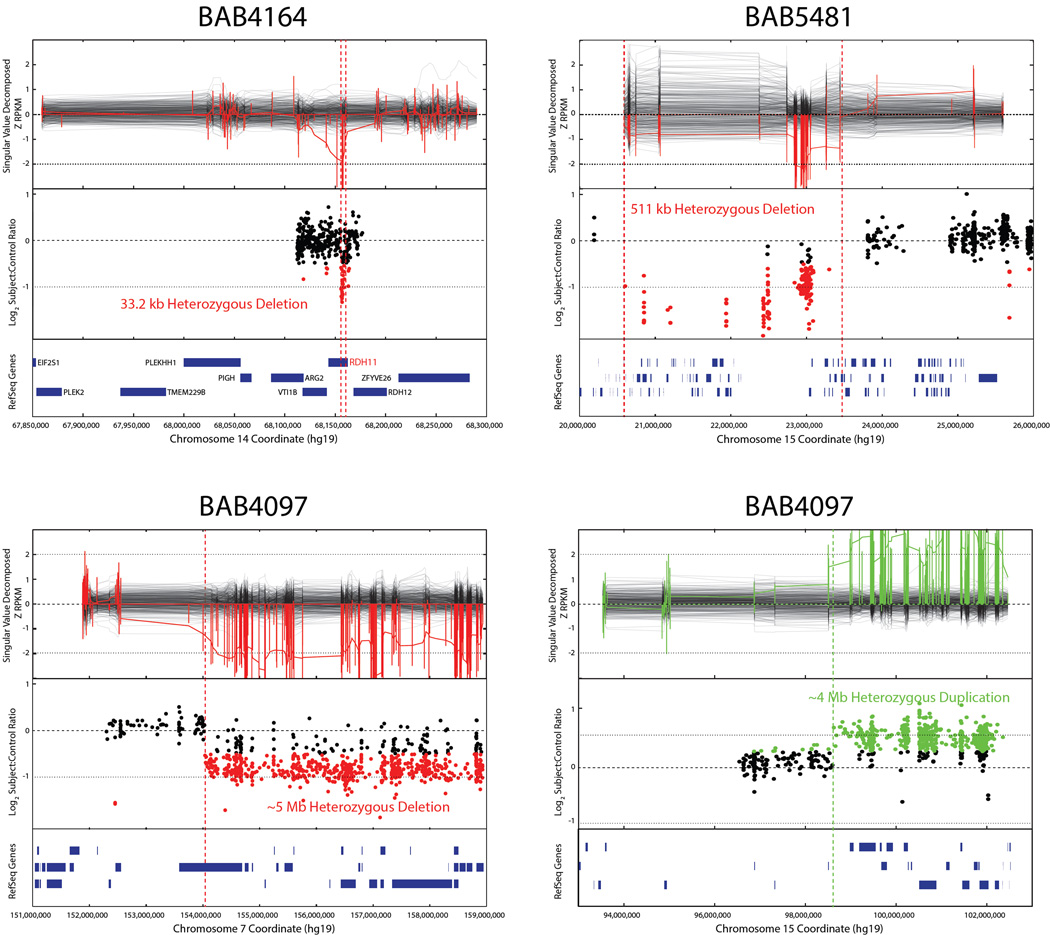
References
-
- Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, Hijazi H, Alshammari M, Aldahmesh MA, Salih MA, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell reports. 2015;10:148–161. - PubMed
-
- Aravind L, Koonin EV. A novel family of predicted phosphoesterases includes Drosophila prune protein and bacterial RecJ exonuclease. Trends in biochemical sciences. 1998;23:17–19. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
