

High-throughput identification of protein mutant stability computed from a double mutant fitness landscape
- PMID: 26540565
- PMCID: PMC4815338
- DOI: 10.1002/pro.2840
High-throughput identification of protein mutant stability computed from a double mutant fitness landscape
Abstract
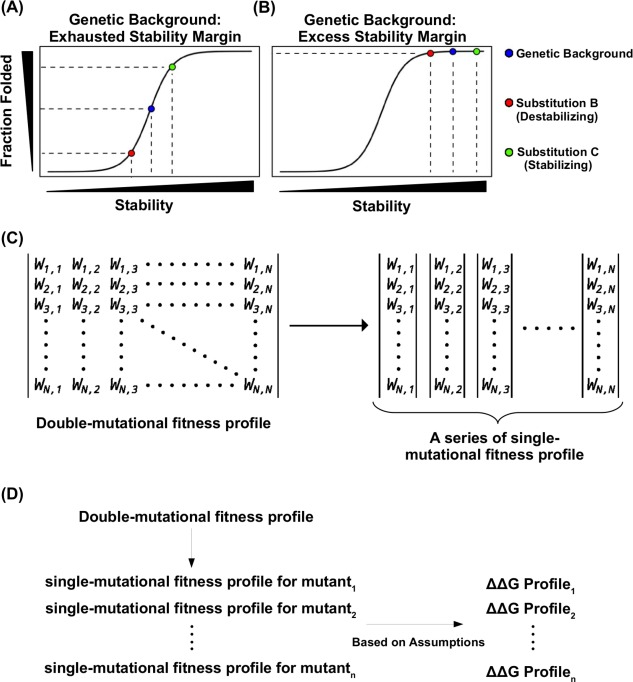
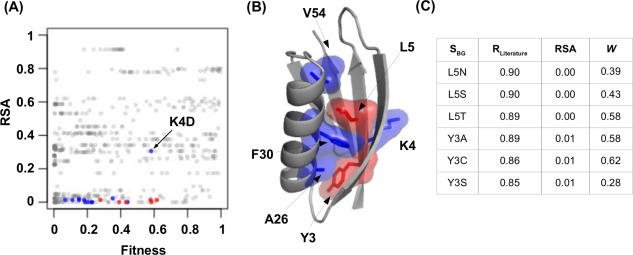
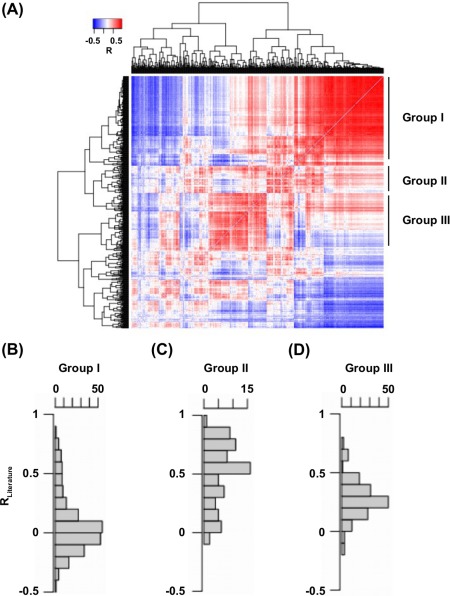
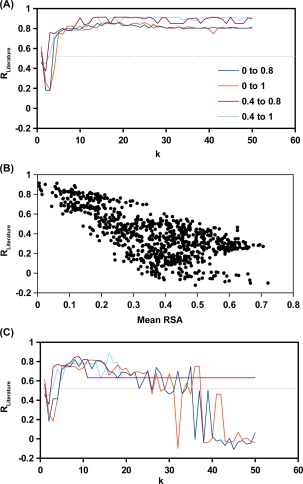
The effect of a mutation on protein stability is traditionally measured by genetic construction, expression, purification, and physical analysis using low-throughput methods. This process is tedious and limits the number of mutants able to be examined in a single study. In contrast, functional fitness effects can be measured in a high-throughput manner by various deep mutational scanning tools. Using protein GB 1, we have recently demonstrated the feasibility of estimating the mutational stability effect ( ΔΔG) of single-substitution based on the functional fitness profile of all double-substitutions. The principle is to identify genetic backgrounds that have an exhausted stability margin. The functional effect of an additional substitution on these genetic backgrounds can then be used to compute the mutational ΔΔG based on the biophysical relationship between functional fitness and thermodynamic stability. However, to identify such genetic backgrounds, the approach described in our previous study required a benchmark dataset, which is a set of known mutational ΔΔG. In this study, a benchmark-independent approach is developed. The genetic backgrounds of interest are identified using k-means clustering with the integration of structural information. We further demonstrated that a reasonable approximation of ΔΔG can also be obtained without taking structural information into account. In summary, this study describes a novel method for computing ΔΔG from double-substitution functional fitness profiles alone, without relying on any known mutational ΔΔG as a benchmark.
Keywords: fitness profiling; mutagenesis; mutant stability prediction; protein stability.
© 2015 The Protein Society.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
