

Non-muscle myosin II in disease: mechanisms and therapeutic opportunities
- PMID: 26542704
- PMCID: PMC4728321
- DOI: 10.1242/dmm.022103
Non-muscle myosin II in disease: mechanisms and therapeutic opportunities
Abstract
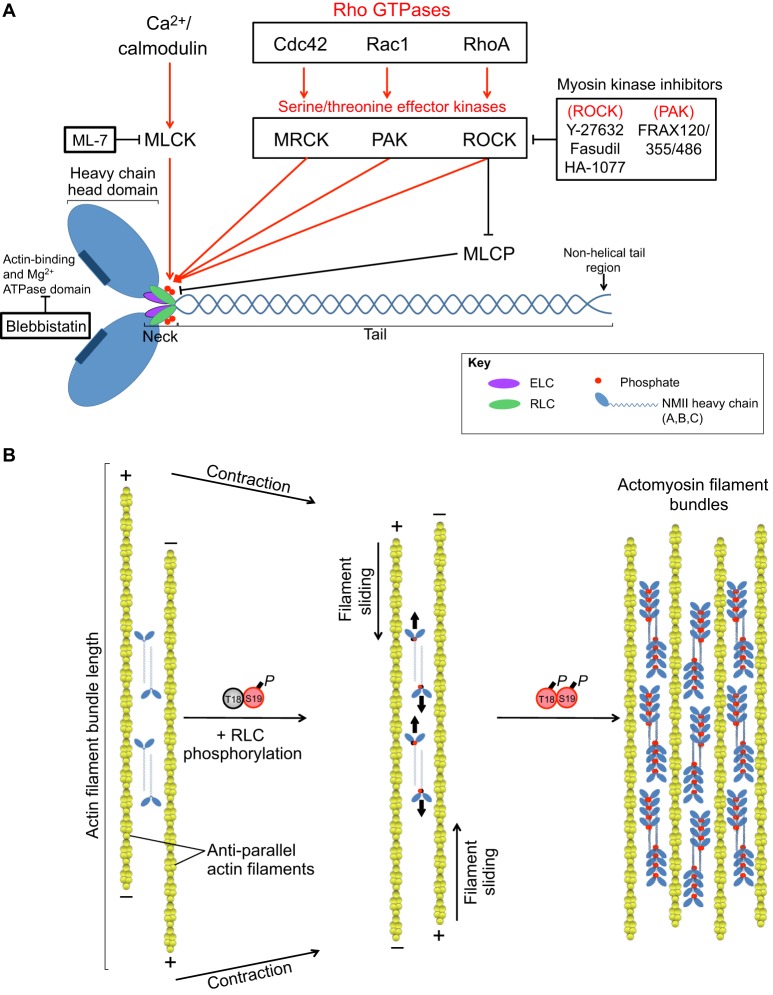
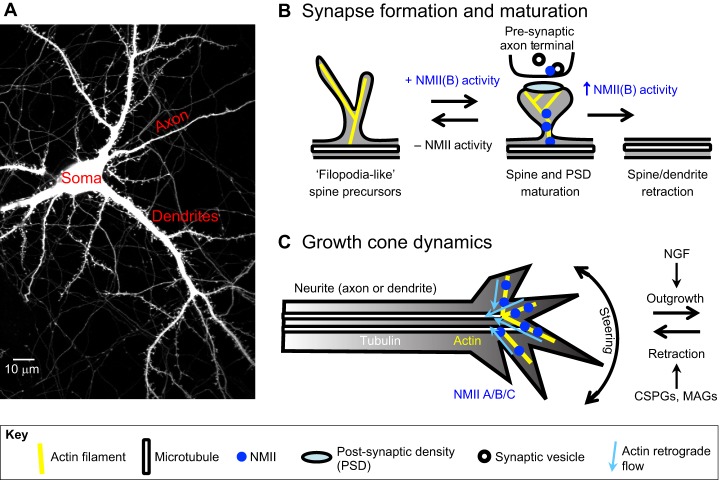
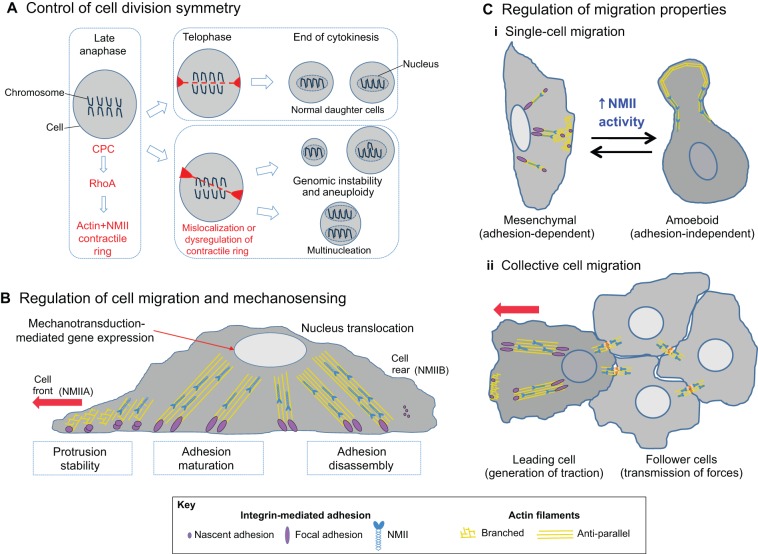
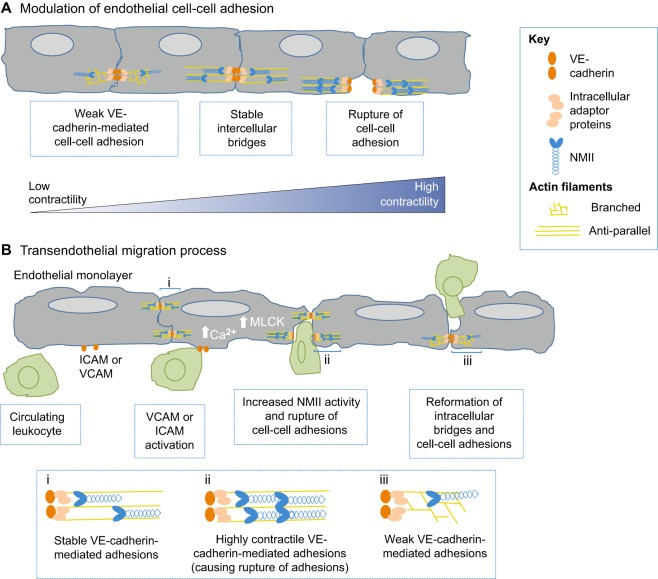
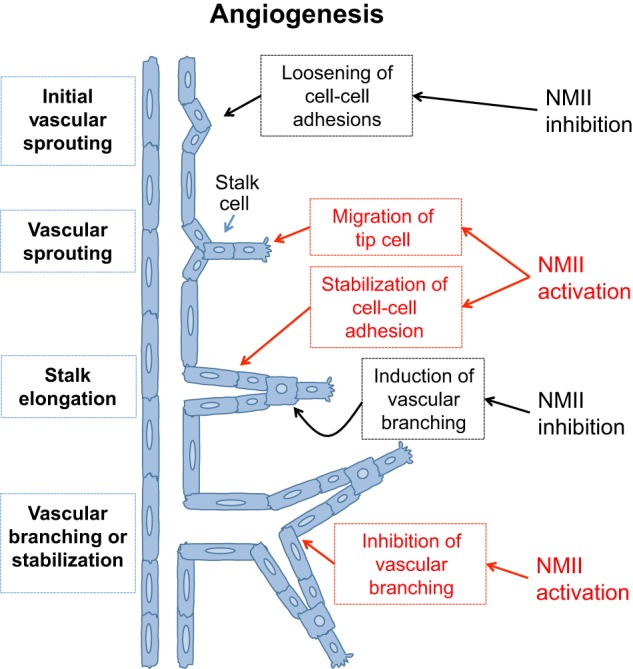
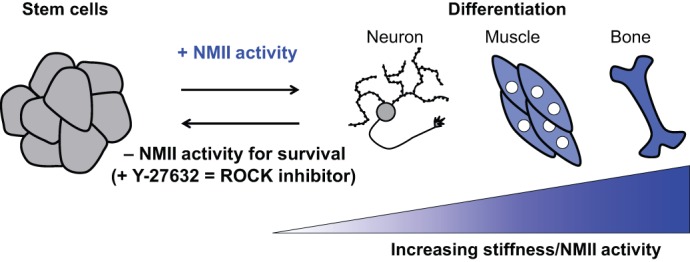
The actin motor protein non-muscle myosin II (NMII) acts as a master regulator of cell morphology, with a role in several essential cellular processes, including cell migration and post-synaptic dendritic spine plasticity in neurons. NMII also generates forces that alter biochemical signaling, by driving changes in interactions between actin-associated proteins that can ultimately regulate gene transcription. In addition to its roles in normal cellular physiology, NMII has recently emerged as a critical regulator of diverse, genetically complex diseases, including neuronal disorders, cancers and vascular disease. In the context of these disorders, NMII regulatory pathways can be directly mutated or indirectly altered by disease-causing mutations. NMII regulatory pathway genes are also increasingly found in disease-associated copy-number variants, particularly in neuronal disorders such as autism and schizophrenia. Furthermore, manipulation of NMII-mediated contractility regulates stem cell pluripotency and differentiation, thus highlighting the key role of NMII-based pharmaceuticals in the clinical success of stem cell therapies. In this Review, we discuss the emerging role of NMII activity and its regulation by kinases and microRNAs in the pathogenesis and prognosis of a diverse range of diseases, including neuronal disorders, cancer and vascular disease. We also address promising clinical applications and limitations of NMII-based inhibitors in the treatment of these diseases and the development of stem-cell-based therapies.
Keywords: Migration; Myosin; NMII; Stem cell; Synapse.
© 2015. Published by The Company of Biologists Ltd.
Conflict of interest statement
The authors declare no competing or financial interests.
Figures
References
-
- Alon U., Barkai N., Notterman D. A., Gish K., Ybarra S., Mack D. and Levine A. J. (1999). Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 96, 6745-6750. 10.1073/pnas.96.12.6745 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
