

Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications
- PMID: 26543054
- PMCID: PMC4674593
- DOI: 10.1530/EJE-15-0515
Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications
Abstract
Objective: Hereditary hypophosphatemias (HH) are rare monogenic conditions characterized by decreased renal tubular phosphate reabsorption. The aim of this study was to explore the prevalence, genotypes, phenotypic spectrum, treatment response, and complications of treatment in the Norwegian population of children with HH.
Design: Retrospective national cohort study.
Methods: Sanger sequencing and multiplex ligand-dependent probe amplification analysis of PHEX and Sanger sequencing of FGF23, DMP1, ENPP1KL, and FAM20C were performed to assess genotype in patients with HH with or without rickets in all pediatric hospital departments across Norway. Patients with hypercalcuria were screened for SLC34A3 mutations. In one family, exome sequencing was performed. Information from the patients' medical records was collected for the evaluation of phenotype.
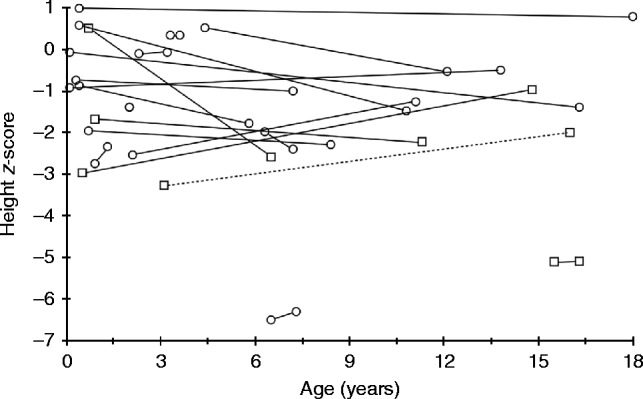
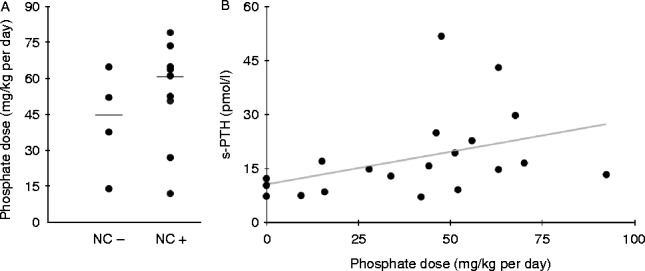
Results: Twety-eight patients with HH (18 females and ten males) from 19 different families were identified. X-linked dominant hypophosphatemic rickets (XLHR) was confirmed in 21 children from 13 families. The total number of inhabitants in Norway aged 18 or below by 1st January 2010 was 1,109,156, giving an XLHR prevalence of ∼1 in 60,000 Norwegian children. FAM20C mutations were found in two brothers and SLC34A3 mutations in one patient. In XLHR, growth was compromised in spite of treatment with oral phosphate and active vitamin D compounds, with males tending to be more affected than females. Nephrocalcinosis tended to be slightly more common in patients starting treatment before 1 year of age, and was associated with higher average treatment doses of phosphate. However, none of these differences reached statistical significance.
Conclusions: We present the first national cohort of HH in children. The prevalence of XLHR seems to be lower in Norwegian children than reported earlier.
© 2016 The authors.
Figures
References
-
- Francis F, Hennign S, Korn B, Reinhardt R, de Jong P, Poustka A, Lehrach H, Rowe PSN, Goulding JN, Summerfield T, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nature Genetics. 1995;11:130–136. doi: 10.1038/ng1095-130. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
