

Monitoring Cellular Phosphorylation Signaling Pathways into Chromatin and Down to the Gene Level
- PMID: 26543102
- PMCID: PMC4813704
- DOI: 10.1074/mcp.M115.053421
Monitoring Cellular Phosphorylation Signaling Pathways into Chromatin and Down to the Gene Level
Abstract
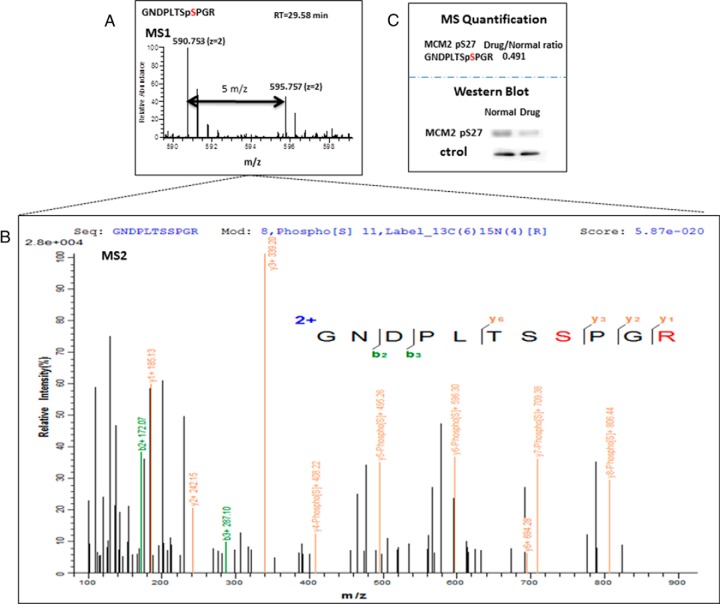
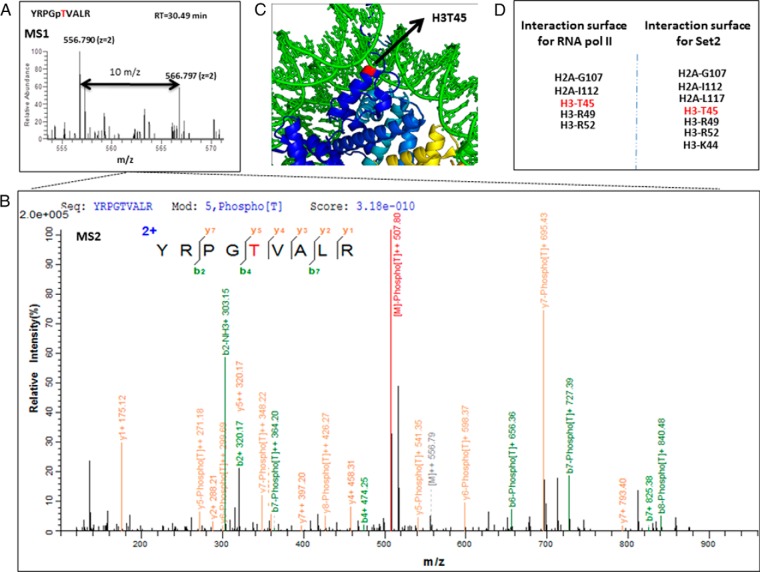
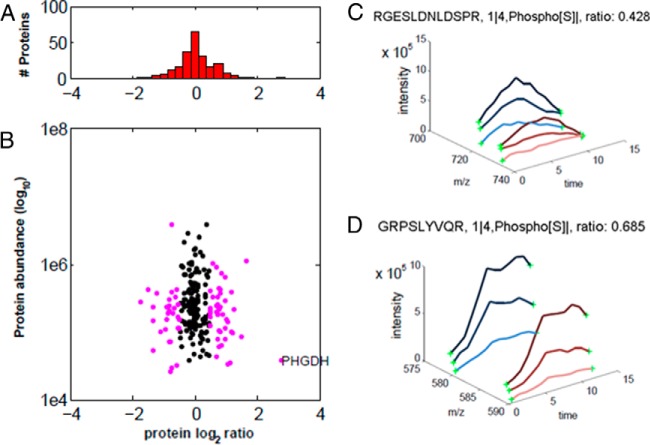
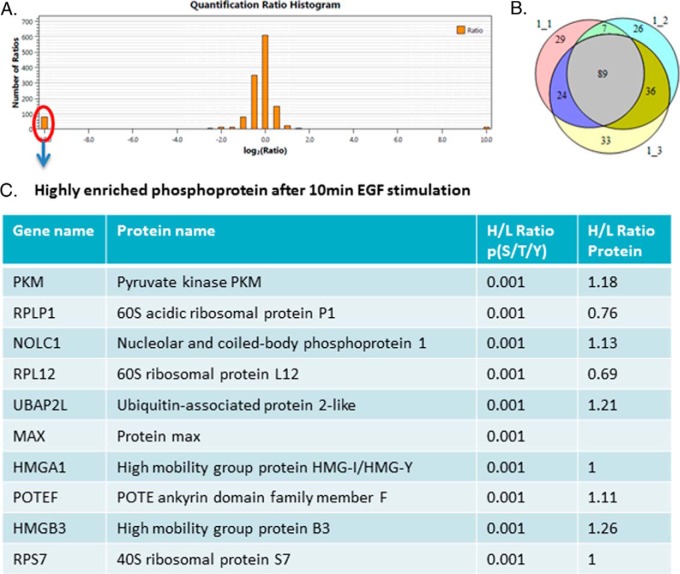
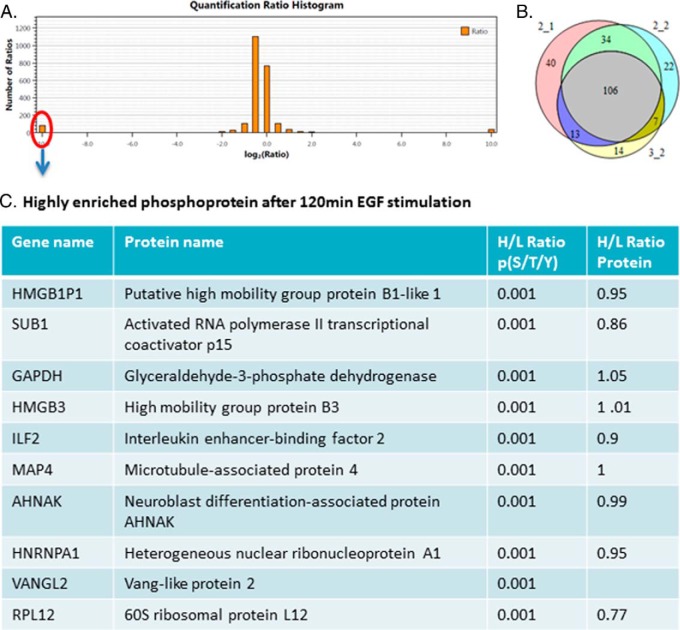
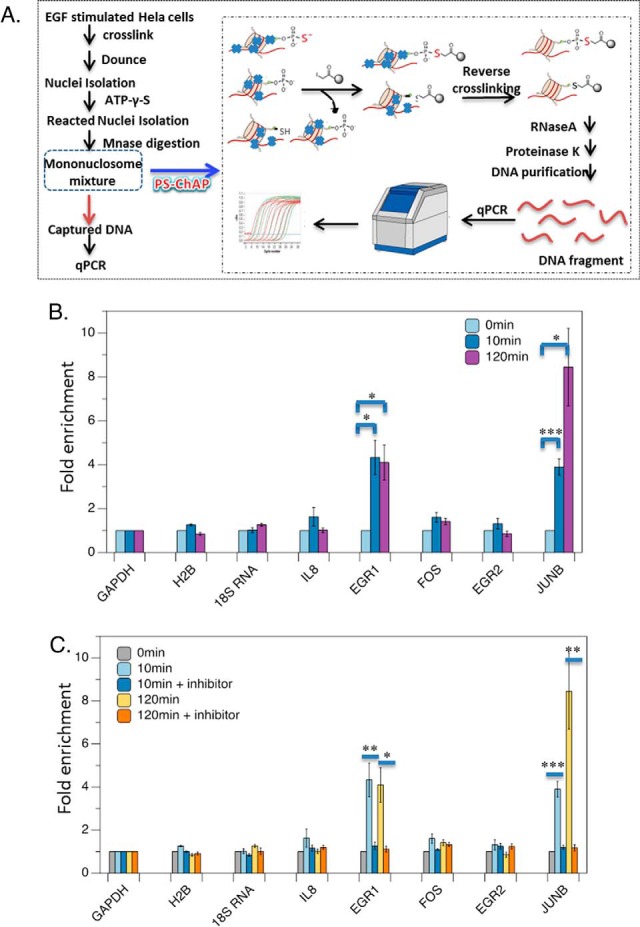
Protein phosphorylation, one of the most common and important modifications of acute and reversible regulation of protein function, plays a dominant role in almost all cellular processes. These signaling events regulate cellular responses, including proliferation, differentiation, metabolism, survival, and apoptosis. Several studies have been successfully used to identify phosphorylated proteins and dynamic changes in phosphorylation status after stimulation. Nevertheless, it is still rather difficult to elucidate precise complex phosphorylation signaling pathways. In particular, how signal transduction pathways directly communicate from the outer cell surface through cytoplasmic space and then directly into chromatin networks to change the transcriptional and epigenetic landscape remains poorly understood. Here, we describe the optimization and comparison of methods based on thiophosphorylation affinity enrichment, which can be utilized to monitor phosphorylation signaling into chromatin by isolation of phosphoprotein containing nucleosomes, a method we term phosphorylation-specific chromatin affinity purification (PS-ChAP). We utilized this PS-ChAP(1) approach in combination with quantitative proteomics to identify changes in the phosphorylation status of chromatin-bound proteins on nucleosomes following perturbation of transcriptional processes. We also demonstrate that this method can be employed to map phosphoprotein signaling into chromatin containing nucleosomes through identifying the genes those phosphorylated proteins are found on via thiophosphate PS-ChAP-qPCR. Thus, our results showed that PS-ChAP offers a new strategy for studying cellular signaling and chromatin biology, allowing us to directly and comprehensively investigate phosphorylation signaling into chromatin to investigate if these pathways are involved in altering gene expression. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the data set identifier PXD002436.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
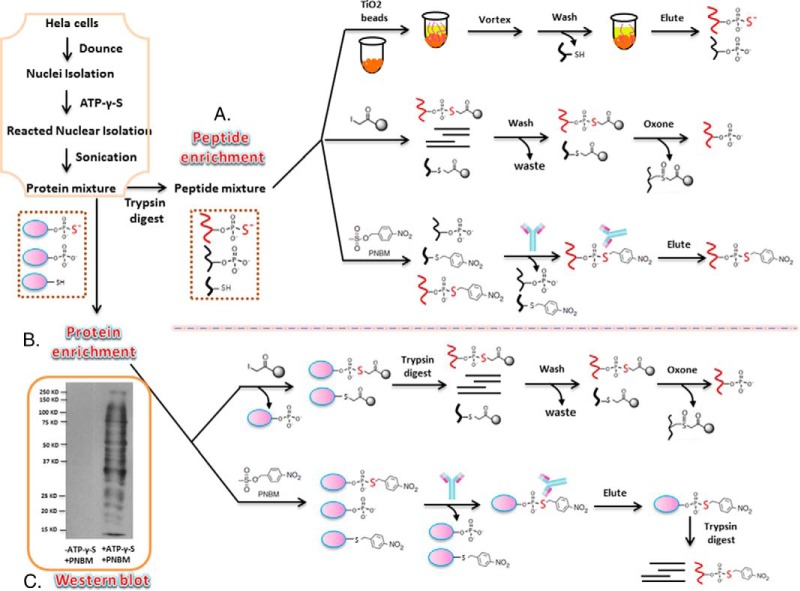
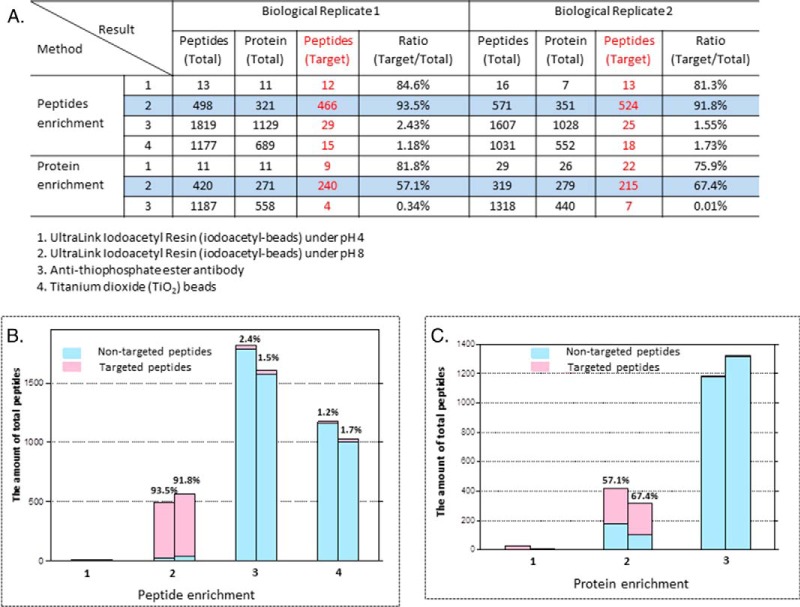
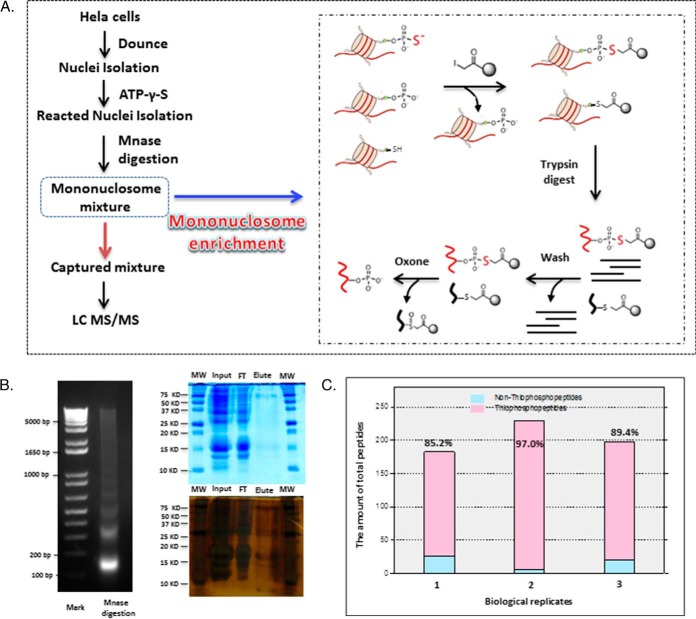
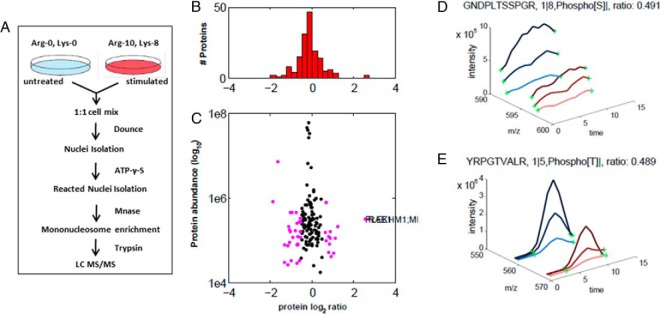
References
-
- Cohen P. (2002) The origins of protein phosphorylation. Nat. Cell Biol. 4, E127–E130 - PubMed
-
- Hunter T. (1995) Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell 80, 225–236 - PubMed
-
- Hunter T. (2000) Signaling—2000 and beyond. Cell 100, 113–127 - PubMed
-
- Cohen P. (2001) The role of protein phosphorylation in human health and disease. Eur. J. Biochem. 268, 5001–5010 - PubMed
-
- Hubbard M. J., and Cohen P. (1993) On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem. Sci. 18, 172–177 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
