

Reproducible quantitative proteotype data matrices for systems biology
- PMID: 26543201
- PMCID: PMC4710225
- DOI: 10.1091/mbc.E15-07-0507
Reproducible quantitative proteotype data matrices for systems biology
Abstract
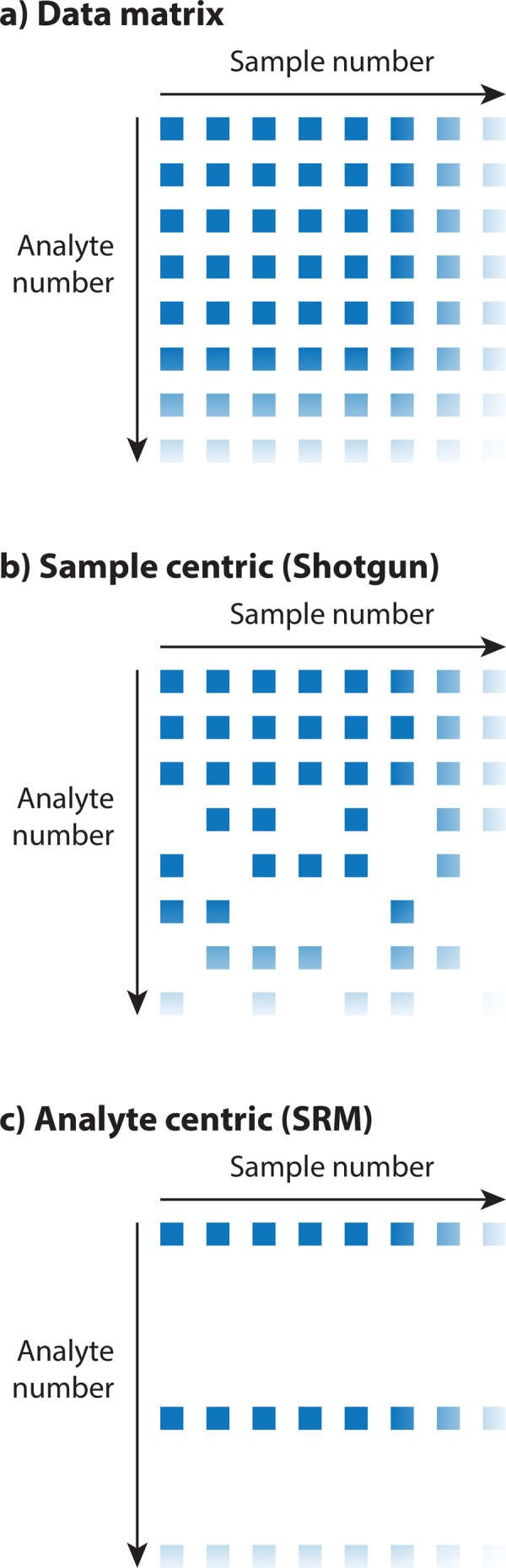
Historically, many mass spectrometry-based proteomic studies have aimed at compiling an inventory of protein compounds present in a biological sample, with the long-term objective of creating a proteome map of a species. However, to answer fundamental questions about the behavior of biological systems at the protein level, accurate and unbiased quantitative data are required in addition to a list of all protein components. Fueled by advances in mass spectrometry, the proteomics field has thus recently shifted focus toward the reproducible quantification of proteins across a large number of biological samples. This provides the foundation to move away from pure enumeration of identified proteins toward quantitative matrices of many proteins measured across multiple samples. It is argued here that data matrices consisting of highly reproducible, quantitative, and unbiased proteomic measurements across a high number of conditions, referred to here as quantitative proteotype maps, will become the fundamental currency in the field and provide the starting point for downstream biological analysis. Such proteotype data matrices, for example, are generated by the measurement of large patient cohorts, time series, or multiple experimental perturbations. They are expected to have a large effect on systems biology and personalized medicine approaches that investigate the dynamic behavior of biological systems across multiple perturbations, time points, and individuals.
© 2015 Röst et al. This article is distributed by The American Society for Cell Biology under license from the author(s). Two months after publication it is available to the public under an Attribution–Noncommercial–Share Alike 3.0 Unported Creative Commons License (http://creativecommons.org/licenses/by-nc-sa/3.0).
Figures
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Bernhardt OM, Selevsek N, Gillet LC, Rinner O, Picotti P, Aebersold R, Reiter L. Spectronaut: a fast and efficient algorithm for MRM-like processing of data independent acquisition (SWATH-MS) data. 60th American Society for Mass Spectrometry Conference 2012. 2012
-
- Bruderer R, Bernhardt OM, Gandhi , Miladinovic´ SM, Cheng LY, Messner S, Ehrenberger T, Zanotelli V, Butscheid Y, Escher C, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteomics. 2015;14:1400–1410. - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
