

DNA Repair Cofactors ATMIN and NBS1 Are Required to Suppress T Cell Activation
- PMID: 26544571
- PMCID: PMC4636180
- DOI: 10.1371/journal.pgen.1005645
DNA Repair Cofactors ATMIN and NBS1 Are Required to Suppress T Cell Activation
Abstract
Proper development of the immune system is an intricate process dependent on many factors, including an intact DNA damage response. The DNA double-strand break signaling kinase ATM and its cofactor NBS1 are required during T cell development and for the maintenance of genomic stability. The role of a second ATM cofactor, ATMIN (also known as ASCIZ) in T cells is much less clear, and whether ATMIN and NBS1 function in synergy in T cells is unknown. Here, we investigate the roles of ATMIN and NBS1, either alone or in combination, using murine models. We show loss of NBS1 led to a developmental block at the double-positive stage of T cell development, as well as reduced TCRα recombination, that was unexpectedly neither exacerbated nor alleviated by concomitant loss of ATMIN. In contrast, loss of both ATMIN and NBS1 enhanced DNA damage that drove spontaneous peripheral T cell hyperactivation, proliferation as well as excessive production of proinflammatory cytokines and chemokines, leading to a highly inflammatory environment. Intriguingly, the disease causing T cells were largely proficient for both ATMIN and NBS1. In vivo this resulted in severe intestinal inflammation, colitis and premature death. Our findings reveal a novel model for an intestinal bowel disease phenotype that occurs upon combined loss of the DNA repair cofactors ATMIN and NBS1.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
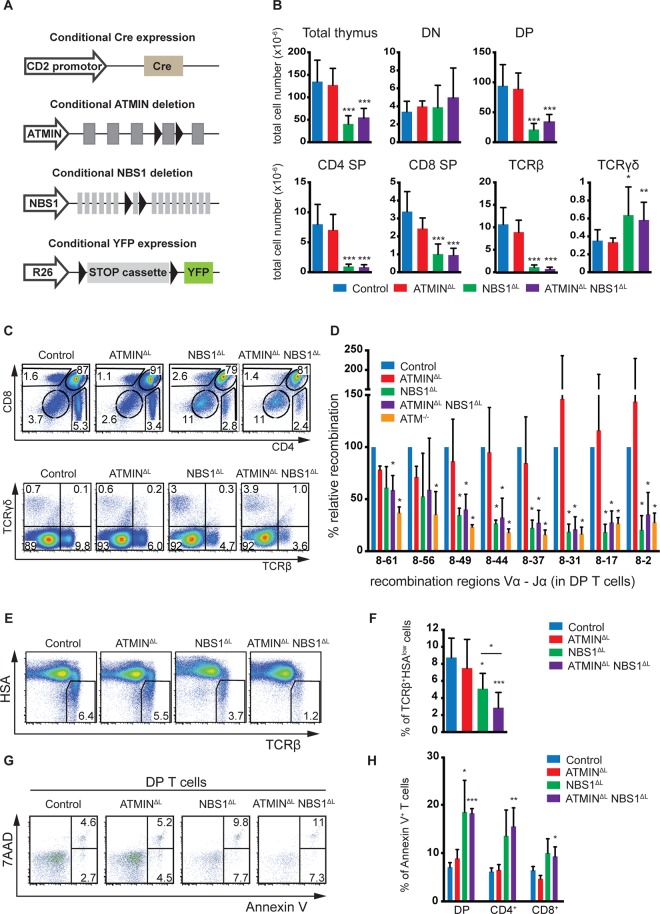
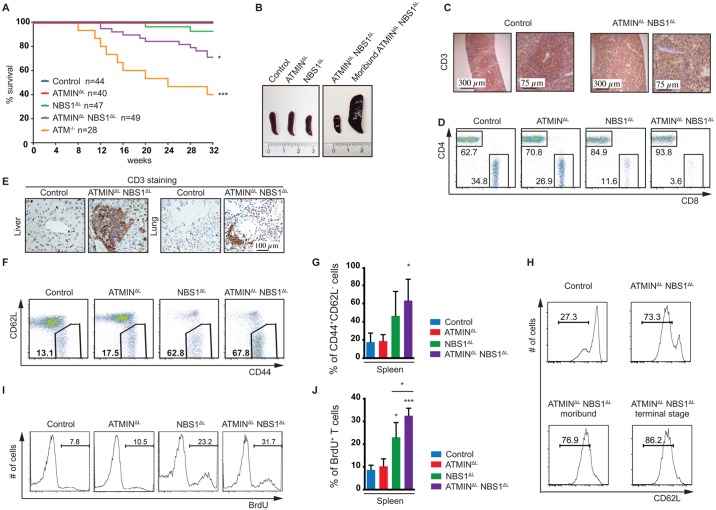
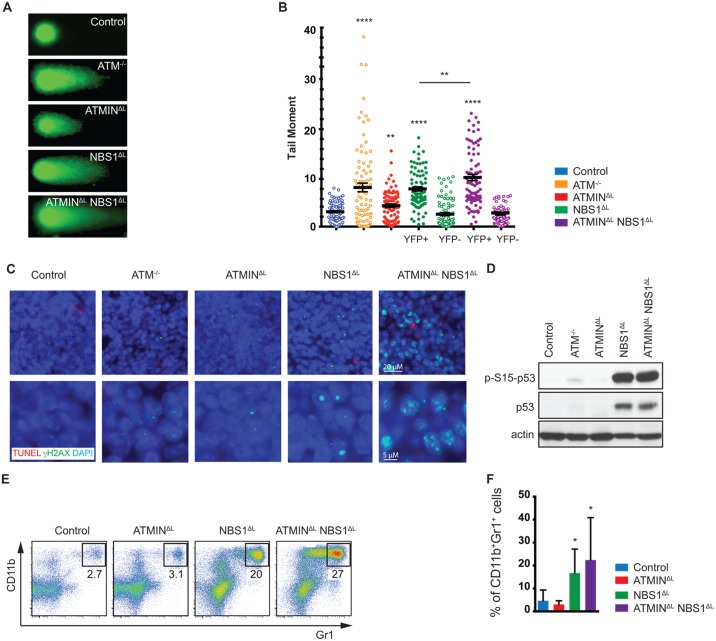
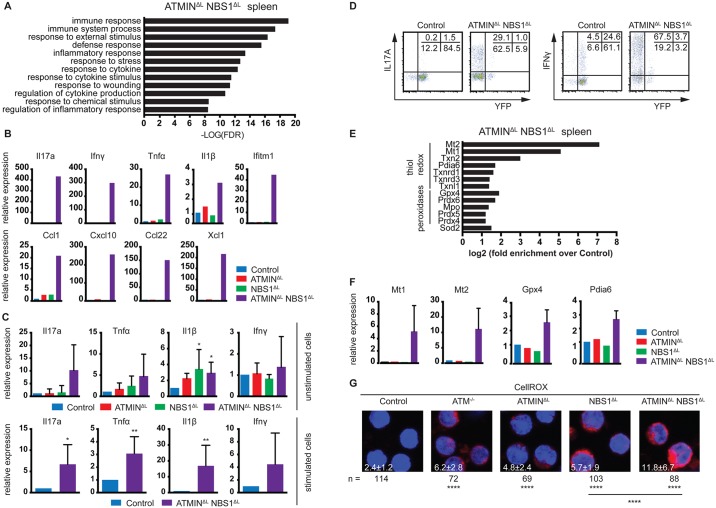
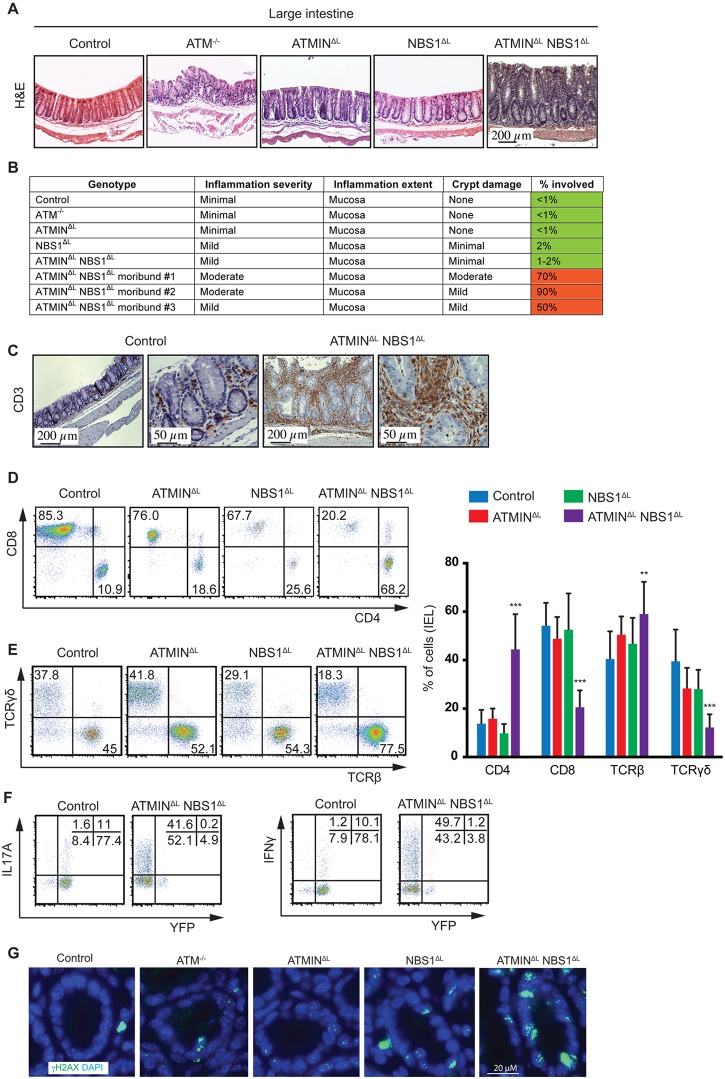
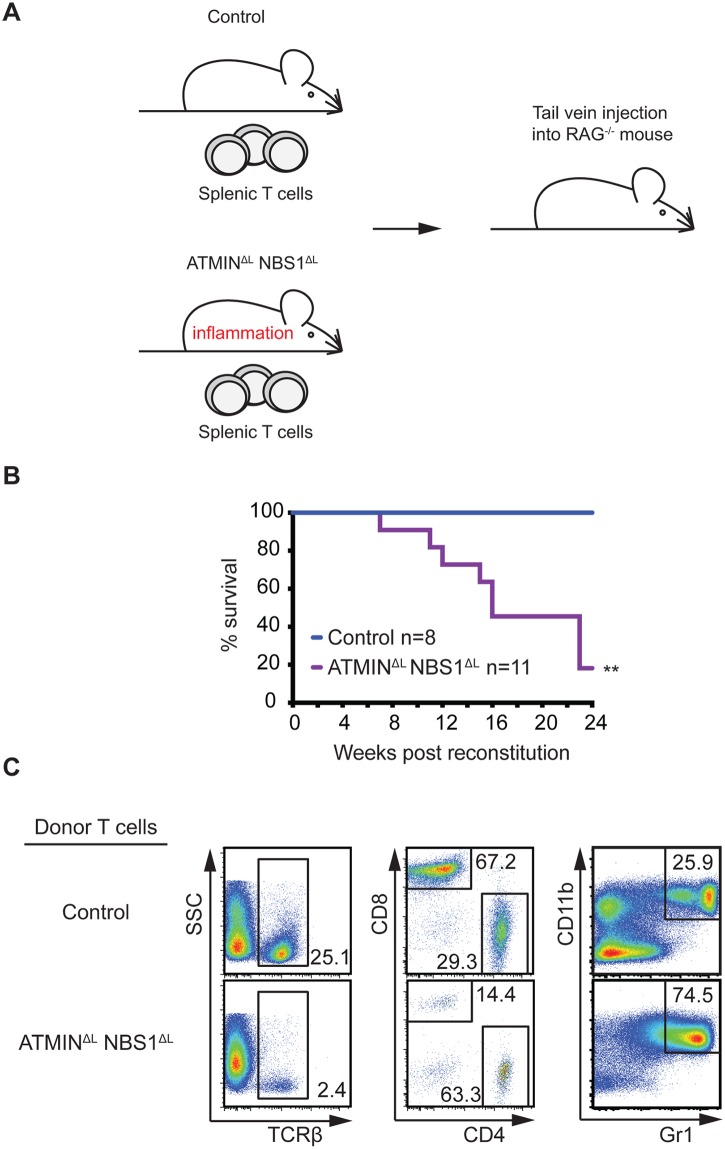

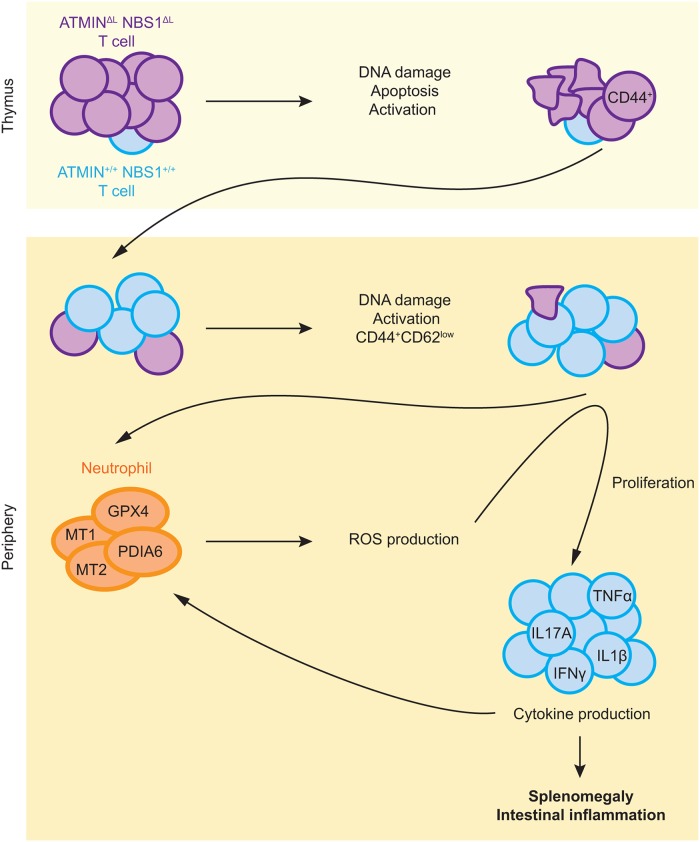
References
-
- Dudley DD, Chaudhuri J, Bassing CH, Alt FW (2005) Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol 86: 43–112. - PubMed
-
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, et al. (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159–171. - PubMed
-
- Callen E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, et al. (2007) ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 130: 63–75. - PubMed
-
- Camacho E, Hernandez L, Hernandez S, Tort F, Bellosillo B, et al. (2002) ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood 99: 238–244. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
