

Computationally Efficient Composite Likelihood Statistics for Demographic Inference
- PMID: 26545922
- PMCID: PMC5854098
- DOI: 10.1093/molbev/msv255
Computationally Efficient Composite Likelihood Statistics for Demographic Inference
Abstract
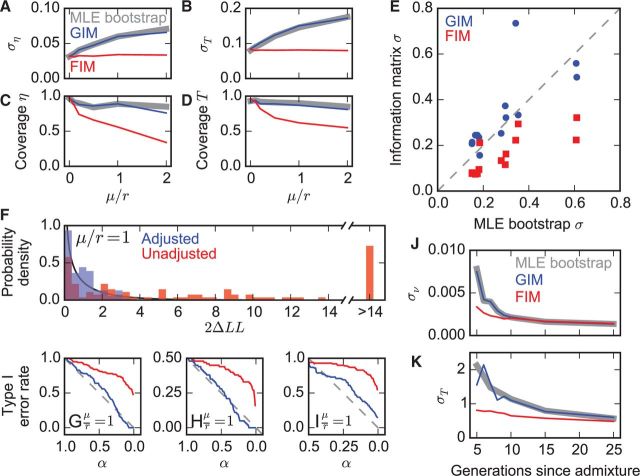
Many population genetics tools employ composite likelihoods, because fully modeling genomic linkage is challenging. But traditional approaches to estimating parameter uncertainties and performing model selection require full likelihoods, so these tools have relied on computationally expensive maximum-likelihood estimation (MLE) on bootstrapped data. Here, we demonstrate that statistical theory can be applied to adjust composite likelihoods and perform robust computationally efficient statistical inference in two demographic inference tools: ∂a∂i and TRACTS. On both simulated and real data, the adjustments perform comparably to MLE bootstrapping while using orders of magnitude less computational time.
Keywords: composite likelihood; demographic inference; likelihood ratio test; parameter uncertainties.
© The Author 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Catelan M, Sartori N. 2015. Empirical and simulated adjustments of composite likelihood ratio statistics. J Stat Comput Simul.
-
- Efron B, Hinkley DV. 1978. Assessing the accuracy of the maximum likelihood estimator: observed versus expected Fisher information. Biometrika 65(3):457–483.
-
- Fearnhead P, Yu S, Biggs P, Holland B, French N. 2015. Estimating the relative rate of recombination to mutation in bacteria from single-locus variants using composite likelihood methods. Ann Appl Stat. 9(1):200–224.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
