

The Redox State Regulates the Conformation of Rv2466c to Activate the Antitubercular Prodrug TP053
- PMID: 26546681
- PMCID: PMC4692232
- DOI: 10.1074/jbc.M115.677039
The Redox State Regulates the Conformation of Rv2466c to Activate the Antitubercular Prodrug TP053
Abstract
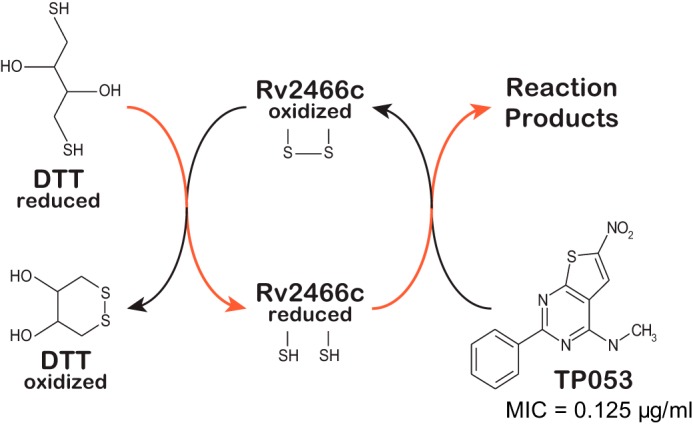
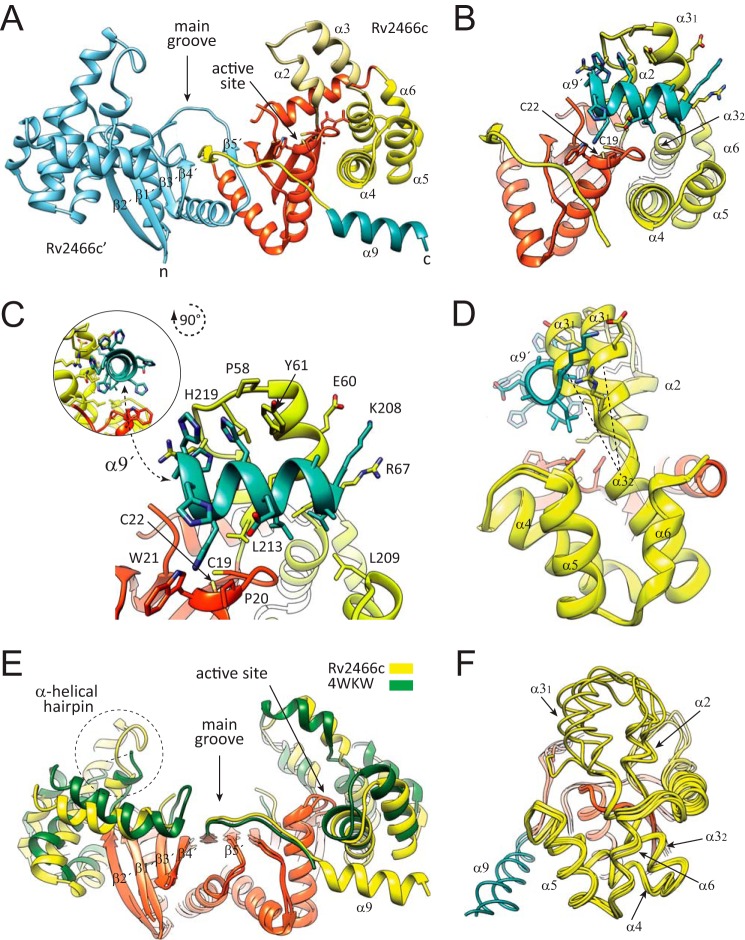
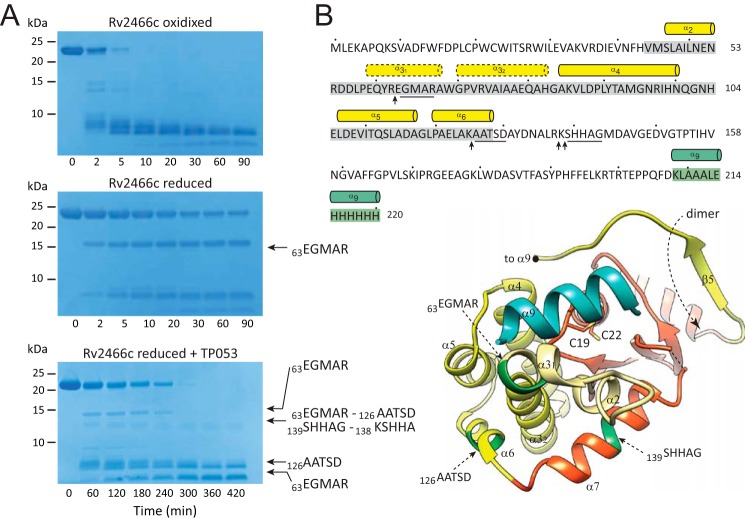
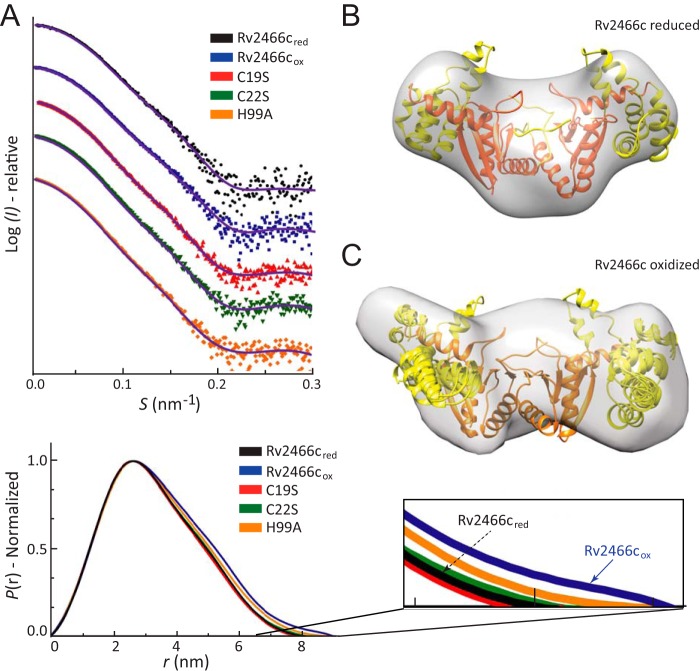
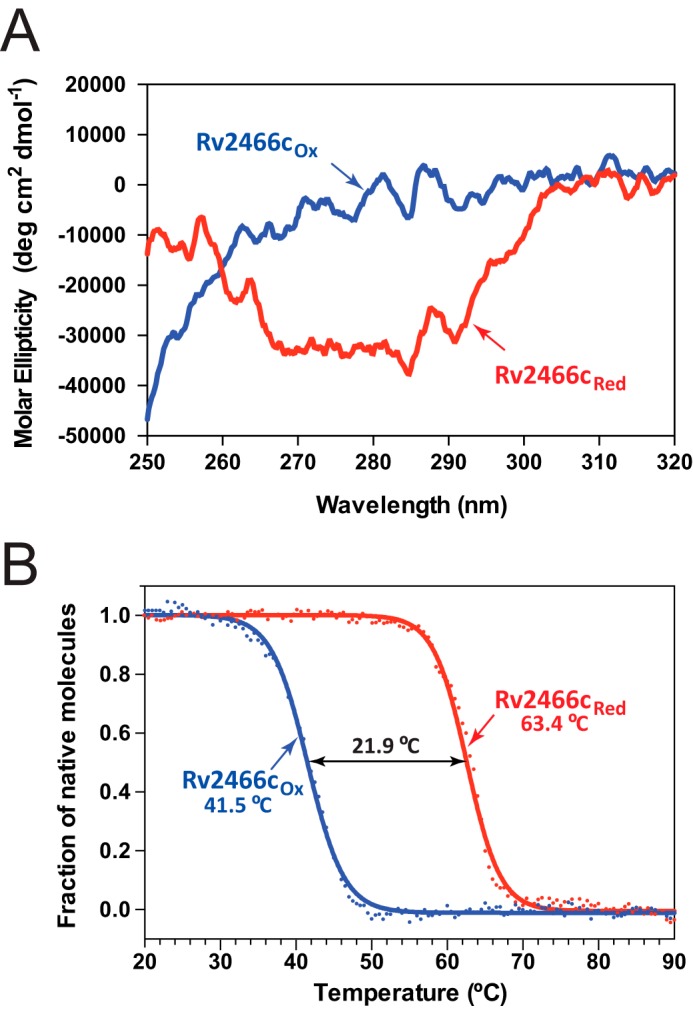
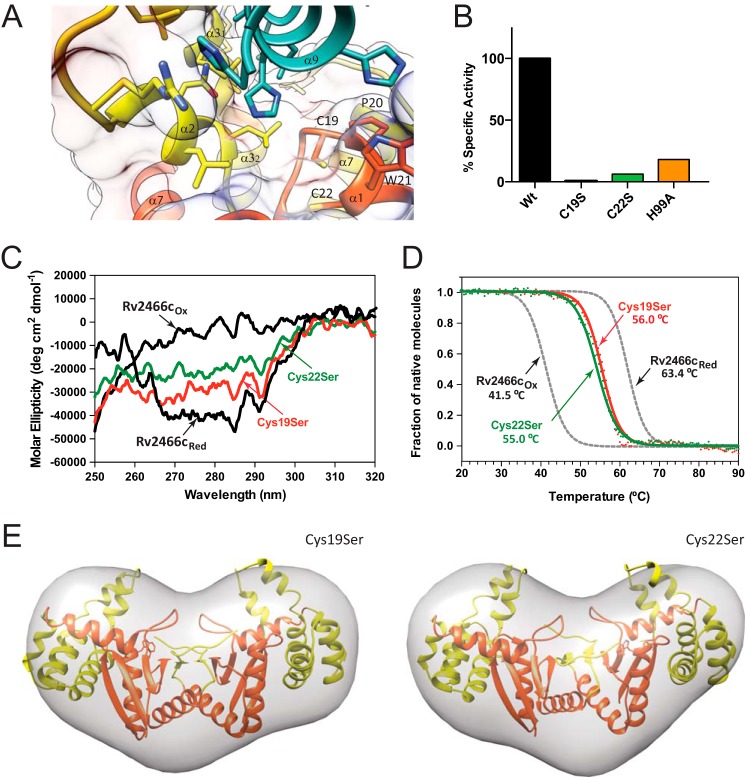
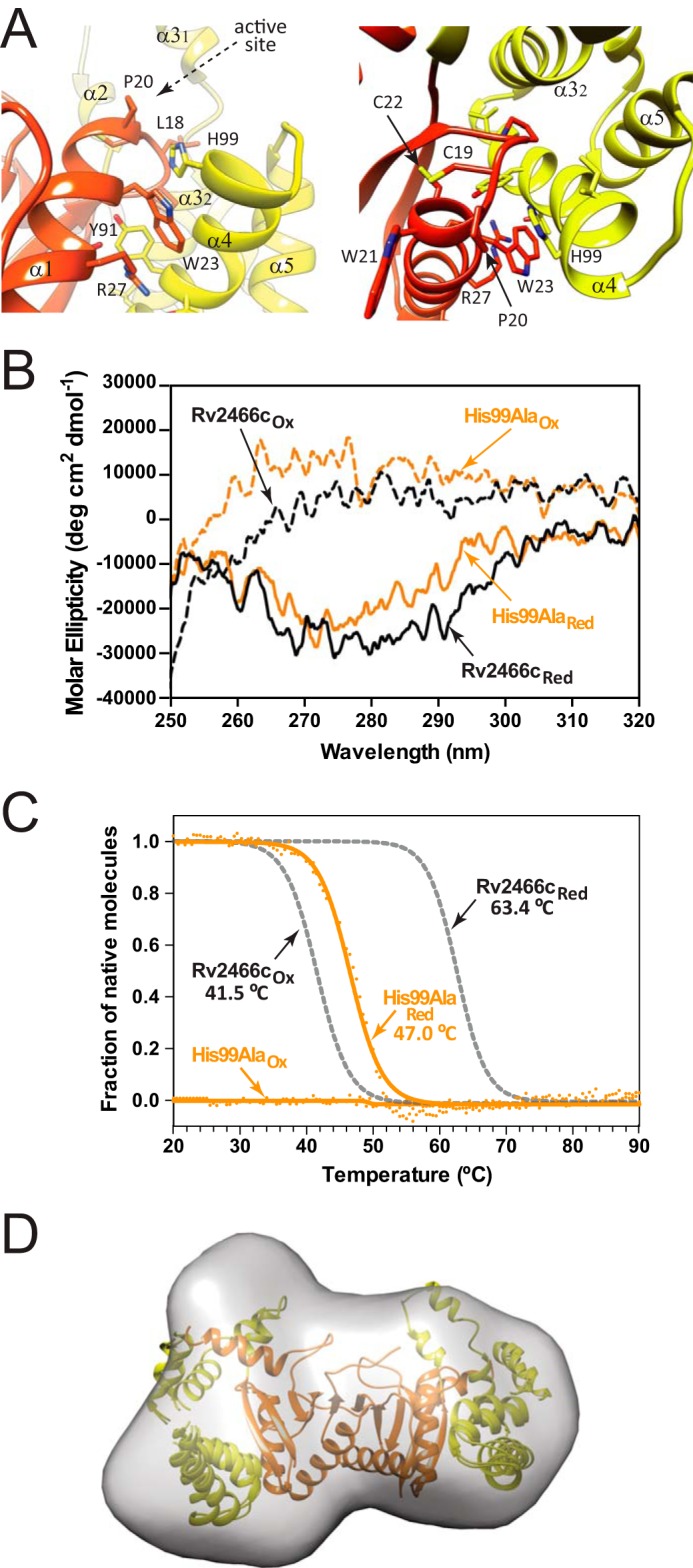
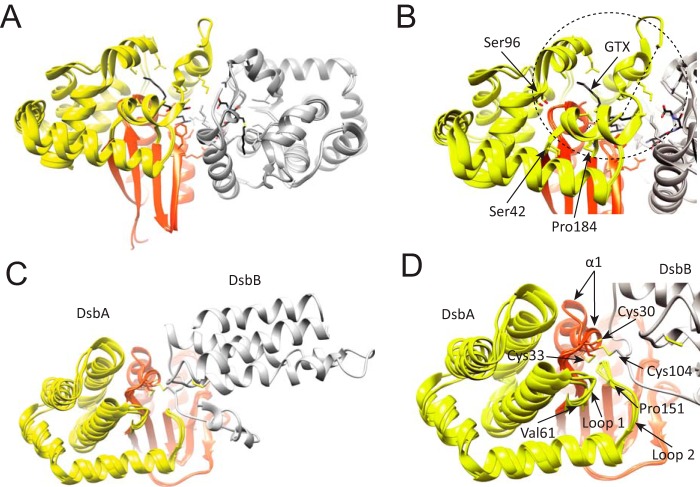
Rv2466c is a key oxidoreductase that mediates the reductive activation of TP053, a thienopyrimidine derivative that kills replicating and non-replicating Mycobacterium tuberculosis, but whose mode of action remains enigmatic. Rv2466c is a homodimer in which each subunit displays a modular architecture comprising a canonical thioredoxin-fold with a Cys(19)-Pro(20)-Trp(21)-Cys(22) motif, and an insertion consisting of a four α-helical bundle and a short α-helical hairpin. Strong evidence is provided for dramatic conformational changes during the Rv2466c redox cycle, which are essential for TP053 activity. Strikingly, a new crystal structure of the reduced form of Rv2466c revealed the binding of a C-terminal extension in α-helical conformation to a pocket next to the active site cysteine pair at the interface between the thioredoxin domain and the helical insertion domain. The ab initio low-resolution envelopes obtained from small angle x-ray scattering showed that the fully reduced form of Rv2466c adopts a "closed" compact conformation in solution, similar to that observed in the crystal structure. In contrast, the oxidized form of Rv2466c displays an "open" conformation, where tertiary structural changes in the α-helical subdomain suffice to account for the observed conformational transitions. Altogether our structural, biochemical, and biophysical data strongly support a model in which the formation of the catalytic disulfide bond upon TP053 reduction triggers local structural changes that open the substrate binding site of Rv2466c allowing the release of the activated, reduced form of TP053. Our studies suggest that similar structural changes might have a functional role in other members of the thioredoxin-fold superfamily.
Keywords: Mycobacterium tuberculosis; conformational change; enzyme; oxidation-reduction (redox); small-angle x-ray scattering (SAXS); thioredoxin; x-ray crystallography.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- World Health Organization (2014) Global Tuberculosis Report 2013, www.who.int/tb/publications/global_report/en
-
- Zumla A., Raviglione M., Hafner R., and von Reyn C. F. (2013) Tuberculosis. N. Engl. J. Med. 368, 745–755 - PubMed
-
- Andries K., Verhasselt P., Guillemont J., Göhlmann H. W., Neefs J. M., Winkler H., Van Gestel J., Timmerman P., Zhu M., Lee E., Williams P., de Chaffoy D., Huitric E., Hoffner S., Cambau E., Truffot-Pernot C., Lounis N., and Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227 - PubMed
-
- Koul A., Dendouga N., Vergauwen K., Molenberghs B., Vranckx L., Willebrords R., Ristic Z., Lill H., Dorange I., Guillemont J., Bald D., and Andries K. (2007) Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 3, 323–324 - PubMed
-
- Cohen J. (2013) Infectious disease: approval of novel TB drug celebrated with restraint. Science 339, 130. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
