

Evidence of Mitochondrial Dysfunction within the Complex Genetic Etiology of Schizophrenia
- PMID: 26550561
- PMCID: PMC4635522
- DOI: 10.1159/000441252
Evidence of Mitochondrial Dysfunction within the Complex Genetic Etiology of Schizophrenia
Erratum in
-
Erratum.Mol Neuropsychiatry. 2016 May;2(1):60. doi: 10.1159/000444029. Epub 2016 May 17. Mol Neuropsychiatry. 2016. PMID: 27602361 Free PMC article.
Abstract
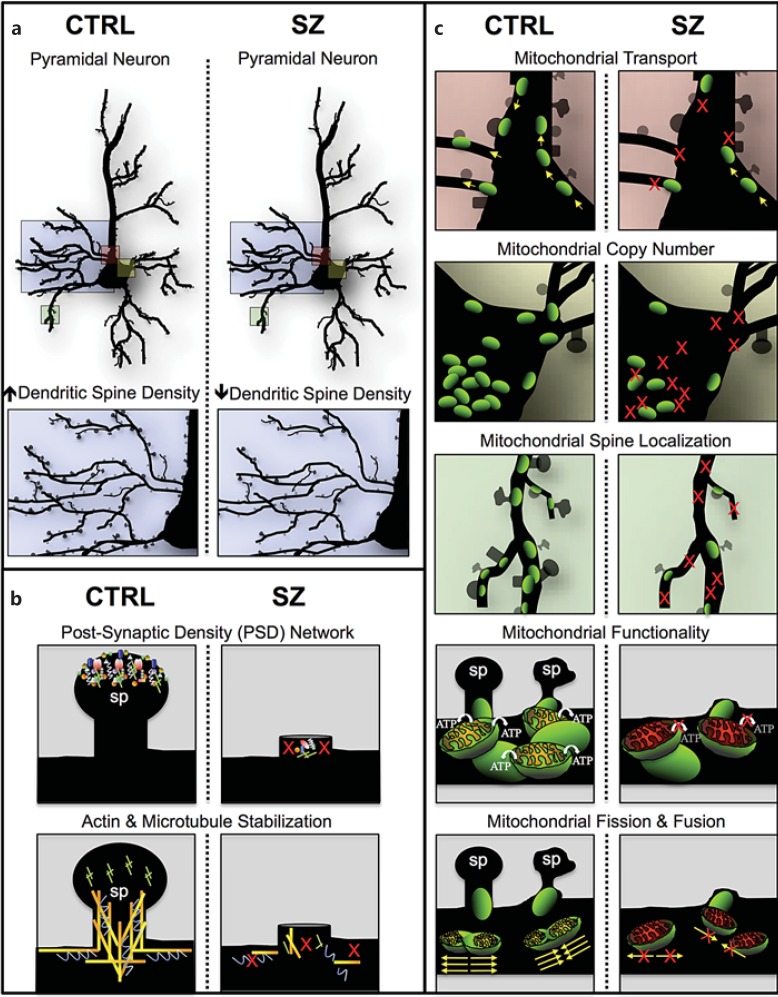
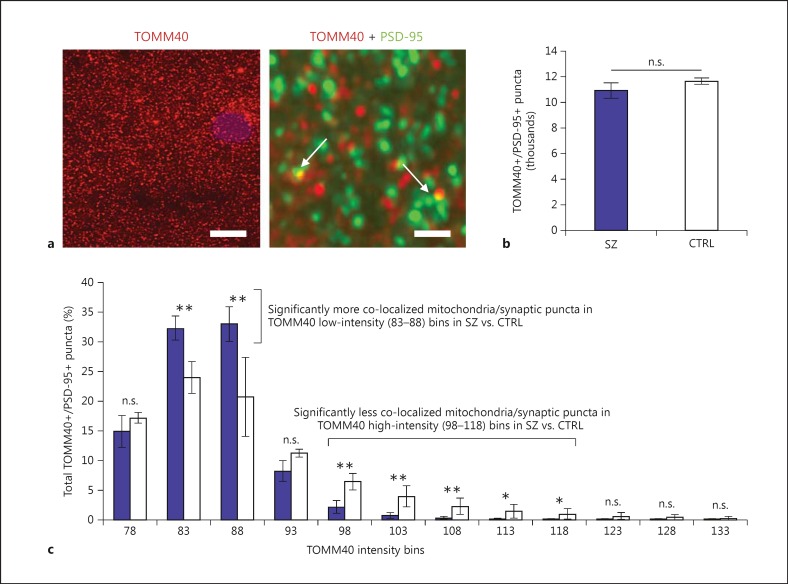
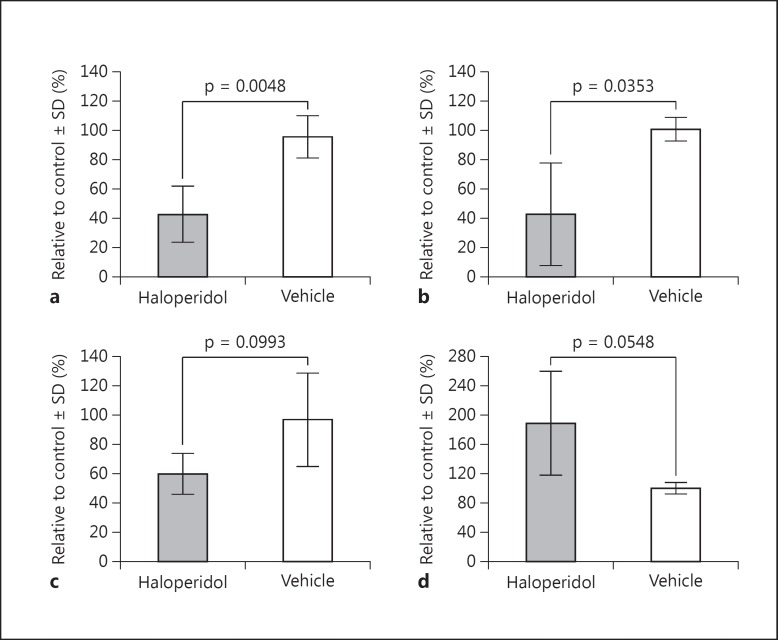
Genetic evidence has supported the hypothesis that schizophrenia (SZ) is a polygenic disorder caused by the disruption in function of several or many genes. The most common and reproducible cellular phenotype associated with SZ is a reduction in dendritic spines within the neocortex, suggesting alterations in dendritic architecture may cause aberrant cortical circuitry and SZ symptoms. Here, we review evidence supporting a multifactorial model of mitochondrial dysfunction in SZ etiology and discuss how these multiple paths to mitochondrial dysfunction may contribute to dendritic spine loss and/or underdevelopment in some SZ subjects. The pathophysiological role of mitochondrial dysfunction in SZ is based upon genomic analyses of both the mitochondrial genome and nuclear genes involved in mitochondrial function. Previous studies and preliminary data suggest SZ is associated with specific alleles and haplogroups of the mitochondrial genome, and also correlates with a reduction in mitochondrial copy number and an increase in synonymous and nonsynonymous substitutions of mitochondrial DNA. Mitochondrial dysfunction has also been widely implicated in SZ by genome-wide association, exome sequencing, altered gene expression, proteomics, microscopy analyses, and induced pluripotent stem cell studies. Together, these data support the hypothesis that SZ is a polygenic disorder with an enrichment of mitochondrial targets.
Keywords: Antipsychotic drug; Dendritic spines; Fluorescence deconvolution tomography; Genome; Induced pluripotent stem cell; Mitochondria; Polygenic disorder; Proteome; Schizophrenia; Transcriptome.
Figures
References
-
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. - PubMed
-
- Katsel P, Davis KL, Gorman JM, Haroutunian V. Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizophr Res. 2005;77:241–252. - PubMed
-
- Vawter MP, Barrett T, Cheadle C, Sokolov BP, Wood WH 3rd, Donovan DM, Webster M, Freed WJ, Becker KG. Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain Res Bull. 2001;55:641–650. - PubMed
