

SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2
- PMID: 26552009
- PMCID: PMC4886303
- DOI: 10.1038/nm.3968
SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2
Abstract
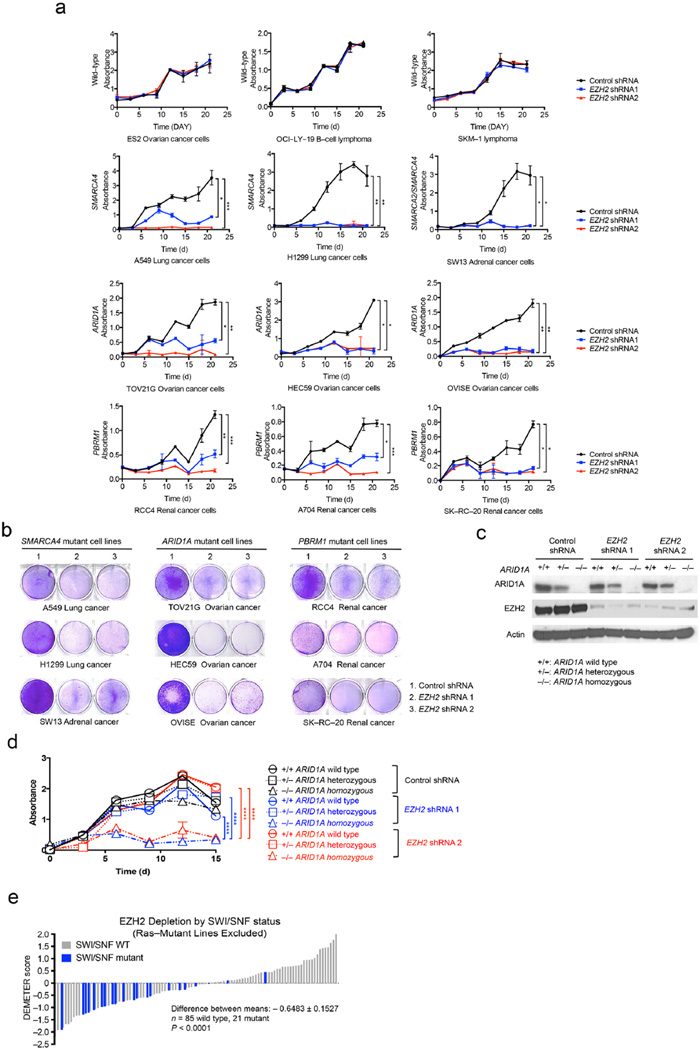
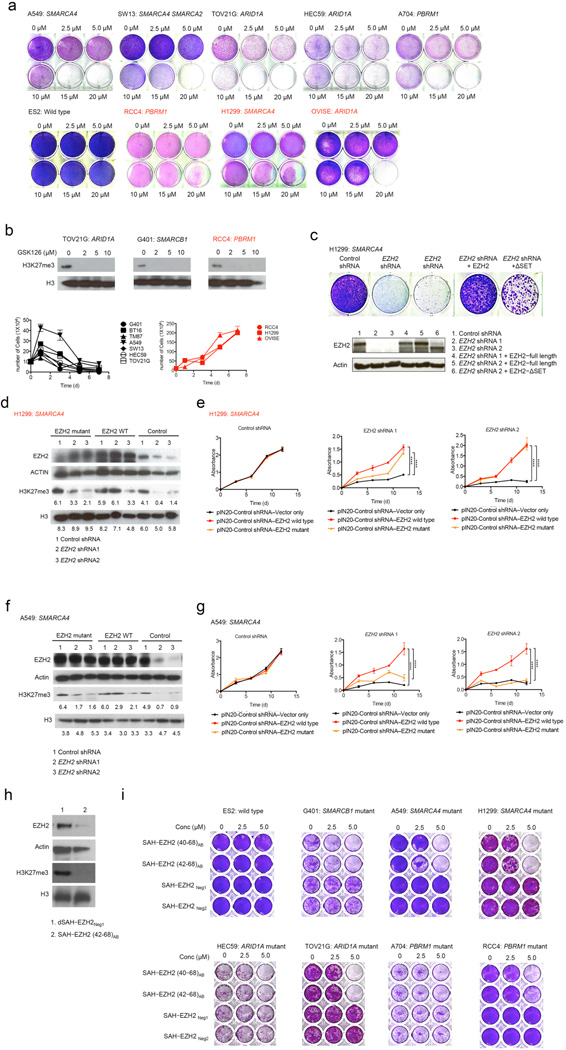
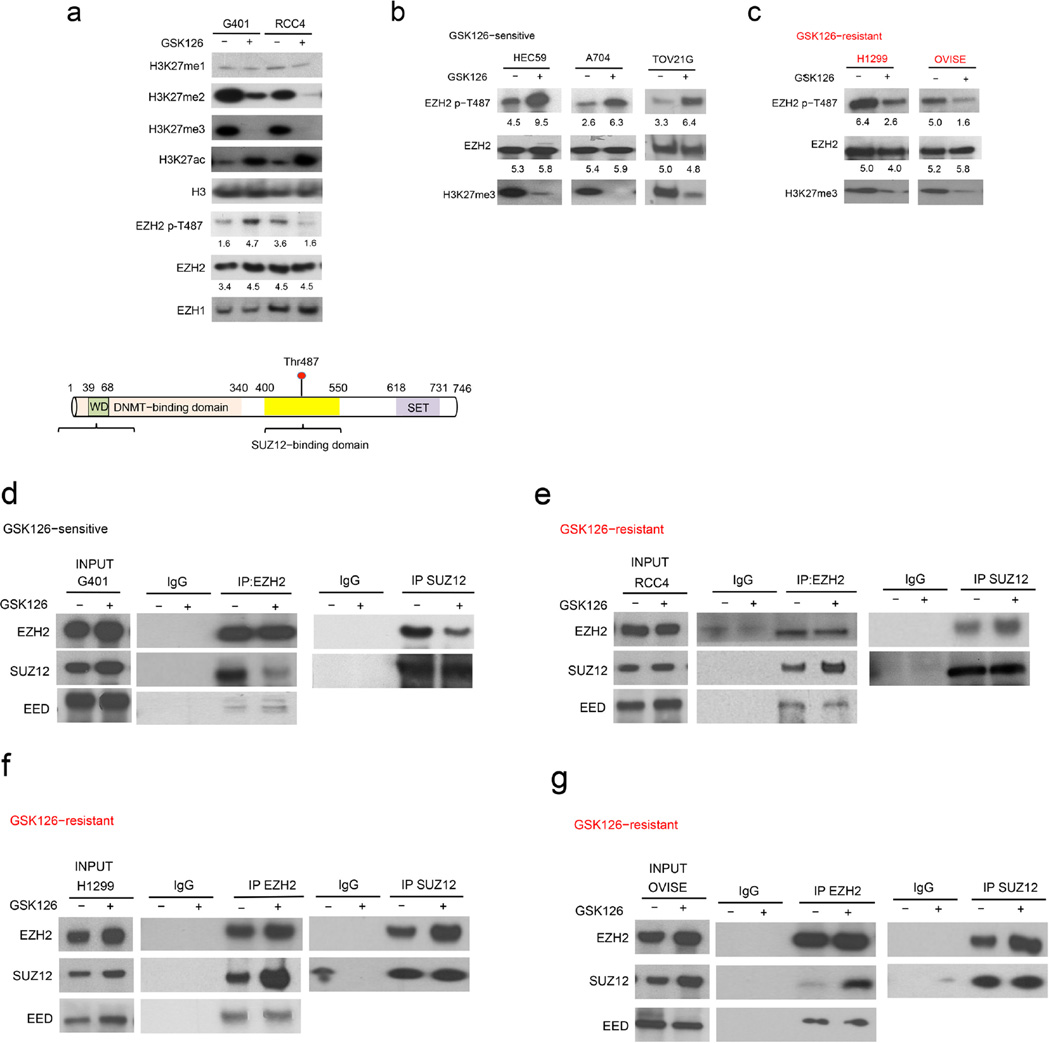
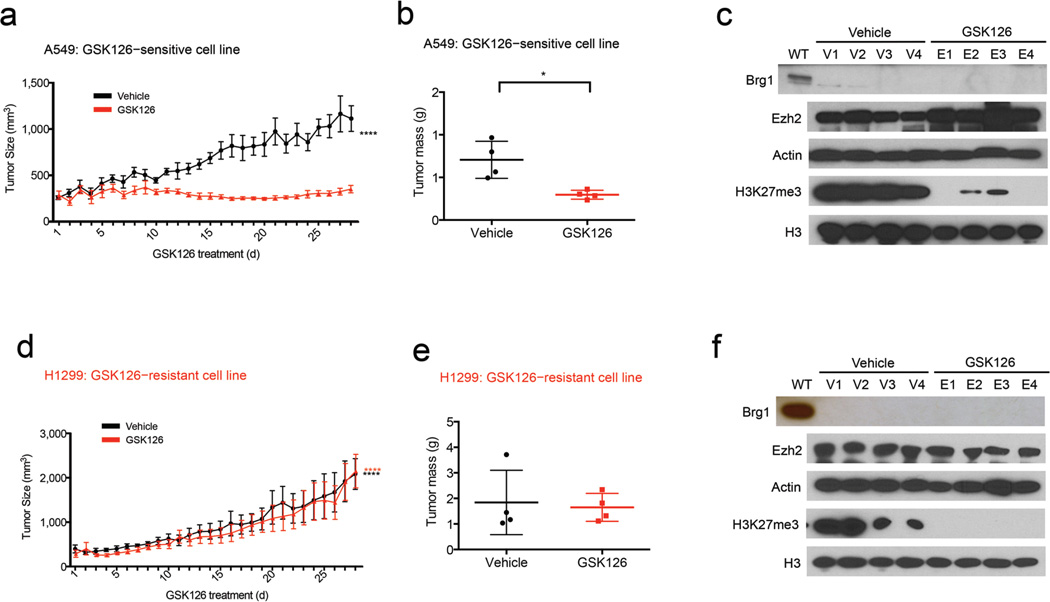
Human cancer genome sequencing has recently revealed that genes that encode subunits of SWI/SNF chromatin remodeling complexes are frequently mutated across a wide variety of cancers, and several subunits of the complex have been shown to have bona fide tumor suppressor activity. However, whether mutations in SWI/SNF subunits result in shared dependencies is unknown. Here we show that EZH2, a catalytic subunit of the polycomb repressive complex 2 (PRC2), is essential in all tested cancer cell lines and xenografts harboring mutations of the SWI/SNF subunits ARID1A, PBRM1, and SMARCA4, which are several of the most frequently mutated SWI/SNF subunits in human cancer, but that co-occurrence of a Ras pathway mutation is correlated with abrogation of this dependence. Notably, we demonstrate that SWI/SNF-mutant cancer cells are primarily dependent on a non-catalytic role of EZH2 in the stabilization of the PRC2 complex, and that they are only partially dependent on EZH2 histone methyltransferase activity. These results not only reveal a shared dependency of cancers with genetic alterations in SWI/SNF subunits, but also suggest that EZH2 enzymatic inhibitors now in clinical development may not fully suppress the oncogenic activity of EZH2.
Figures
Similar articles
-
PRC2-mediated repression of SMARCA2 predicts EZH2 inhibitor activity in SWI/SNF mutant tumors.Proc Natl Acad Sci U S A. 2017 Nov 14;114(46):12249-12254. doi: 10.1073/pnas.1703966114. Epub 2017 Oct 30. Proc Natl Acad Sci U S A. 2017. PMID: 29087303 Free PMC article.
-
SWI/SNF catalytic subunits' switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells.Nat Commun. 2018 Oct 8;9(1):4116. doi: 10.1038/s41467-018-06656-6. Nat Commun. 2018. PMID: 30297712 Free PMC article.
-
Potential of enhancer of zeste homolog 2 inhibitors for the treatment of SWI/SNF mutant cancers and tumor microenvironment modulation.Drug Dev Res. 2021 Sep;82(6):730-753. doi: 10.1002/ddr.21796. Epub 2021 Feb 9. Drug Dev Res. 2021. PMID: 33565092 Review.
-
Selective Killing of SMARCA2- and SMARCA4-deficient Small Cell Carcinoma of the Ovary, Hypercalcemic Type Cells by Inhibition of EZH2: In Vitro and In Vivo Preclinical Models.Mol Cancer Ther. 2017 May;16(5):850-860. doi: 10.1158/1535-7163.MCT-16-0678. Epub 2017 Mar 14. Mol Cancer Ther. 2017. PMID: 28292935
-
PRC2 and SWI/SNF Chromatin Remodeling Complexes in Health and Disease.Biochemistry. 2016 Mar 22;55(11):1600-14. doi: 10.1021/acs.biochem.5b01191. Epub 2016 Feb 17. Biochemistry. 2016. PMID: 26836503 Review.
Cited by
-
The Functional Role of PRC2 in Early T-cell Precursor Acute Lymphoblastic Leukemia (ETP-ALL) - Mechanisms and Opportunities.Front Pediatr. 2016 May 18;4:49. doi: 10.3389/fped.2016.00049. eCollection 2016. Front Pediatr. 2016. PMID: 27242978 Free PMC article. Review.
-
From clinical management to personalized medicine: novel therapeutic approaches for ovarian clear cell cancer.J Ovarian Res. 2024 Feb 12;17(1):39. doi: 10.1186/s13048-024-01359-7. J Ovarian Res. 2024. PMID: 38347608 Free PMC article. Review.
-
Targeting histone modifiers in bladder cancer therapy - preclinical and clinical evidence.Nat Rev Urol. 2024 Aug;21(8):495-511. doi: 10.1038/s41585-024-00857-z. Epub 2024 Feb 19. Nat Rev Urol. 2024. PMID: 38374198 Review.
-
Noncanonical EZH2 drives retinoic acid resistance of variant acute promyelocytic leukemias.Blood. 2022 Dec 1;140(22):2358-2370. doi: 10.1182/blood.2022015668. Blood. 2022. PMID: 35984905 Free PMC article.
-
Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity.Trends Cancer. 2017 May;3(5):372-386. doi: 10.1016/j.trecan.2017.04.004. Epub 2017 May 5. Trends Cancer. 2017. PMID: 28718414 Free PMC article. Review.
References
-
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. - PubMed
References for Methods
-
- Fujimoto A, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature genetics. 2012;44:760–764. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
