

Amyloid β-protein oligomers upregulate the β-secretase, BACE1, through a post-translational mechanism involving its altered subcellular distribution in neurons
- PMID: 26552445
- PMCID: PMC4638102
- DOI: 10.1186/s13041-015-0163-5
Amyloid β-protein oligomers upregulate the β-secretase, BACE1, through a post-translational mechanism involving its altered subcellular distribution in neurons
Abstract
Background: β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) is a membrane-bound aspartyl protease that initiates amyloid β-protein (Aβ) generation. Aberrant elevation of BACE1 levels in brains of Alzheimer's disease (AD) patients may involve Aβ. In the present study, we used a neuron culture model system to investigate the effects of Aβ on BACE1 expression as well as the underlying mechanisms.
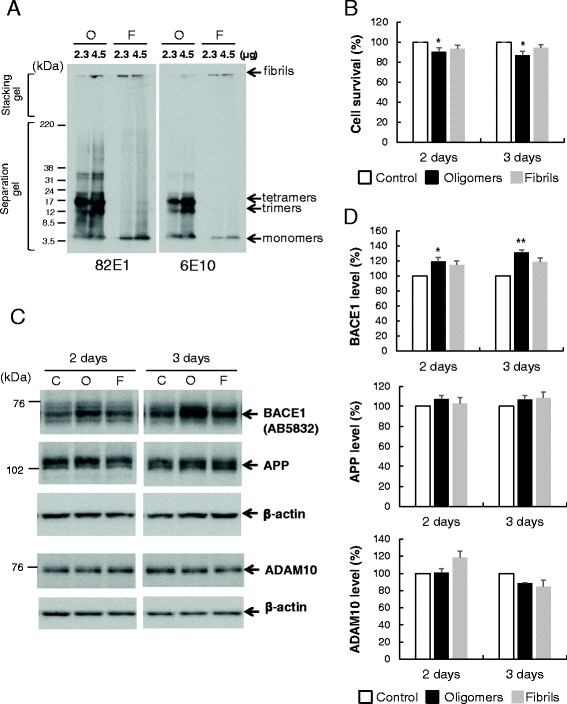
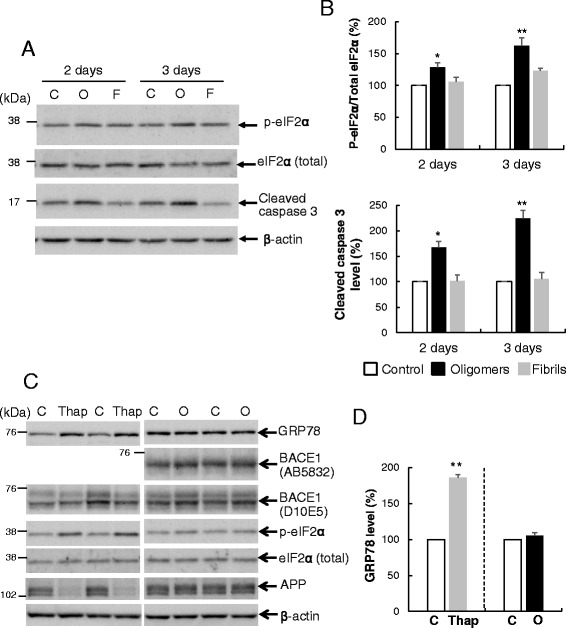
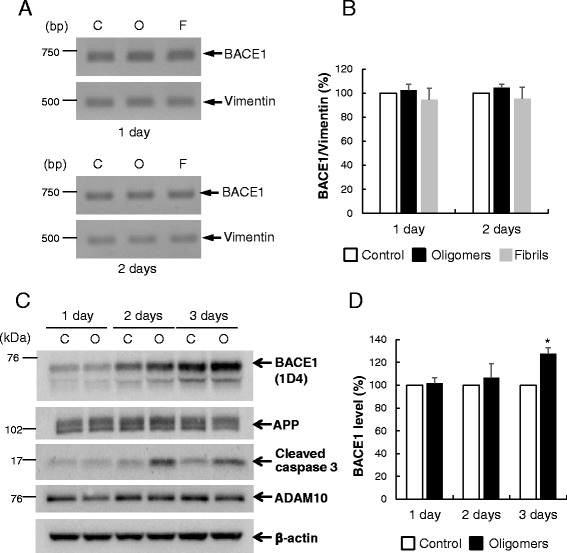
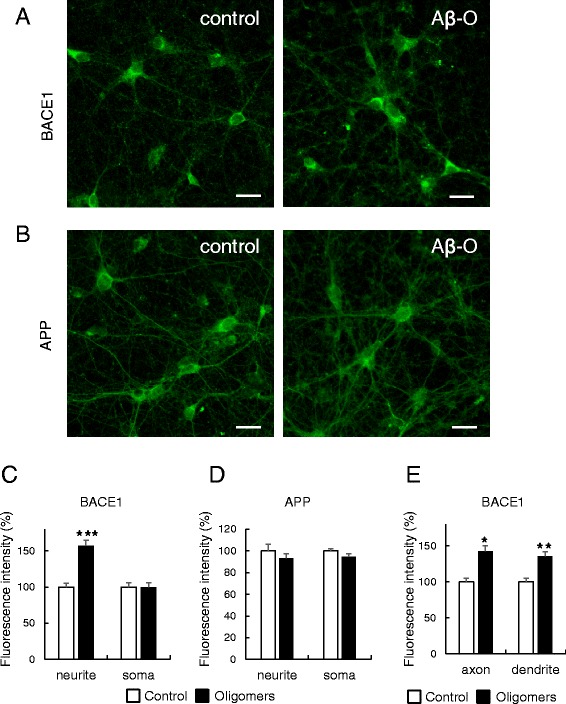
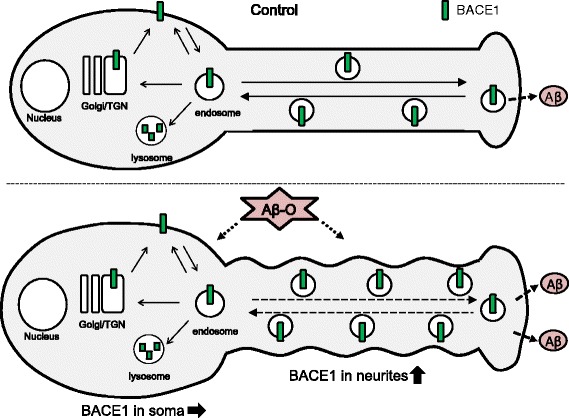
Results: Rat primary cortical neurons were treated with relatively low concentrations (2.5 μM) of Aβ42 oligomers (Aβ-O) or fibrils (Aβ-F) for 2-3 days. Aβ-O induced a significant increase in protein levels of BACE1, while Aβ-F only had a marginal effect. Levels of amyloid precursor protein (APP) and the major α-secretase, ADAM10, remained unaltered upon treatment with both types of Aβ. Aβ-O treatment resulted in activation of eIF2α and caspase 3 in a time-dependent manner, with no changes in the endoplasmic reticulum (ER) stress marker, GRP78, indicating that a typical ER stress response is not induced under our experimental conditions. Furthermore, Aβ-O did not affect BACE1 mRNA expression but augmented the levels of exogenous BACE1 expressed via recombinant adenoviruses, indicating regulation of BACE1 protein expression, not at the transcriptional or translational but the post-translational level. Immunocytochemical analysis revealed that Aβ-O causes a significant increase in BACE1 immunoreactivity in neurites (both axons and dendrites), but not soma of neurons; this change appears relevant to the mechanism of Aβ-O-induced BACE1 elevation, which may involve impairment of BACE1 trafficking and degradation. In contrast, Aβ-O had no effect on APP immunoreactivity.
Conclusion: Our results collectively suggest that Aβ oligomers induce BACE1 elevation via a post-translational mechanism involving its altered subcellular distribution in neurons, which possibly triggers a vicious cycle of Aβ generation, thus contributing to the pathogenetic mechanism of AD.
Figures
Similar articles
-
Genetic inhibition of phosphorylation of the translation initiation factor eIF2α does not block Aβ-dependent elevation of BACE1 and APP levels or reduce amyloid pathology in a mouse model of Alzheimer's disease.PLoS One. 2014 Jul 3;9(7):e101643. doi: 10.1371/journal.pone.0101643. eCollection 2014. PLoS One. 2014. PMID: 24992504 Free PMC article.
-
Regulation of Synaptic Amyloid-β Generation through BACE1 Retrograde Transport in a Mouse Model of Alzheimer's Disease.J Neurosci. 2017 Mar 8;37(10):2639-2655. doi: 10.1523/JNEUROSCI.2851-16.2017. Epub 2017 Feb 3. J Neurosci. 2017. PMID: 28159908 Free PMC article.
-
Relationship between ubiquilin-1 and BACE1 in human Alzheimer's disease and APdE9 transgenic mouse brain and cell-based models.Neurobiol Dis. 2016 Jan;85:187-205. doi: 10.1016/j.nbd.2015.11.005. Epub 2015 Nov 10. Neurobiol Dis. 2016. PMID: 26563932
-
The beta-secretase, BACE: a prime drug target for Alzheimer's disease.J Mol Neurosci. 2001 Oct;17(2):157-70. doi: 10.1385/JMN:17:2:157. J Mol Neurosci. 2001. PMID: 11816789 Review.
-
Function, regulation and therapeutic properties of beta-secretase (BACE1).Semin Cell Dev Biol. 2009 Apr;20(2):175-82. doi: 10.1016/j.semcdb.2009.01.003. Epub 2009 Jan 20. Semin Cell Dev Biol. 2009. PMID: 19429494 Review.
Cited by
-
Pharmacokinetic Modelling of Human Recombinant Protein, p75ECD-Fc: A Novel Therapeutic Approach for Treatment of Alzheimer's Disease, in Serum and Tissue of Sprague Dawley Rats.Eur J Drug Metab Pharmacokinet. 2021 Mar;46(2):235-248. doi: 10.1007/s13318-020-00662-0. Epub 2021 Jan 28. Eur J Drug Metab Pharmacokinet. 2021. PMID: 33507523
-
miR-98-5p Acts as a Target for Alzheimer's Disease by Regulating Aβ Production Through Modulating SNX6 Expression.J Mol Neurosci. 2016 Dec;60(4):413-420. doi: 10.1007/s12031-016-0815-7. Epub 2016 Aug 19. J Mol Neurosci. 2016. PMID: 27541017
-
Astrocytes infected with Chlamydia pneumoniae demonstrate altered expression and activity of secretases involved in the generation of β-amyloid found in Alzheimer disease.BMC Neurosci. 2019 Feb 20;20(1):6. doi: 10.1186/s12868-019-0489-5. BMC Neurosci. 2019. PMID: 30786875 Free PMC article.
-
The neurotoxicity of amyloid β-protein oligomers is reversible in a primary neuron model.Mol Brain. 2017 Jan 31;10(1):4. doi: 10.1186/s13041-016-0284-5. Mol Brain. 2017. PMID: 28137266 Free PMC article.
-
Stable cerebrospinal fluid neurogranin and β-site amyloid precursor protein cleaving enzyme 1 levels differentiate predementia Alzheimer's disease patients.Brain Commun. 2022 Sep 24;4(5):fcac244. doi: 10.1093/braincomms/fcac244. eCollection 2022. Brain Commun. 2022. PMID: 36262371 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
