

Inferring intra-motif dependencies of DNA binding sites from ChIP-seq data
- PMID: 26552868
- PMCID: PMC4640111
- DOI: 10.1186/s12859-015-0797-4
Inferring intra-motif dependencies of DNA binding sites from ChIP-seq data
Abstract
Background: Statistical modeling of transcription factor binding sites is one of the classical fields in bioinformatics. The position weight matrix (PWM) model, which assumes statistical independence among all nucleotides in a binding site, used to be the standard model for this task for more than three decades but its simple assumptions are increasingly put into question. Recent high-throughput sequencing methods have provided data sets of sufficient size and quality for studying the benefits of more complex models. However, learning more complex models typically entails the danger of overfitting, and while model classes that dynamically adapt the model complexity to data have been developed, effective model selection is to date only possible for fully observable data, but not, e.g., within de novo motif discovery.
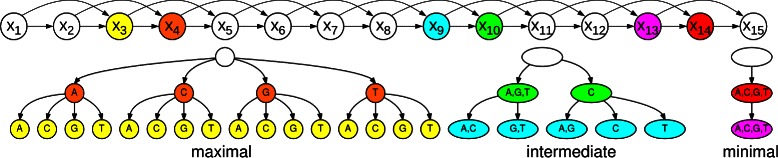
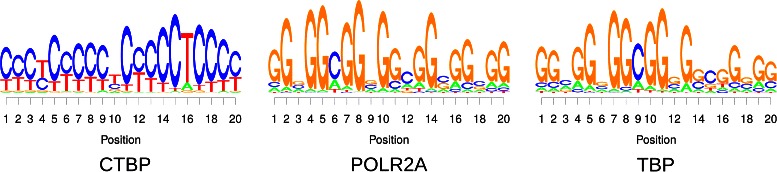
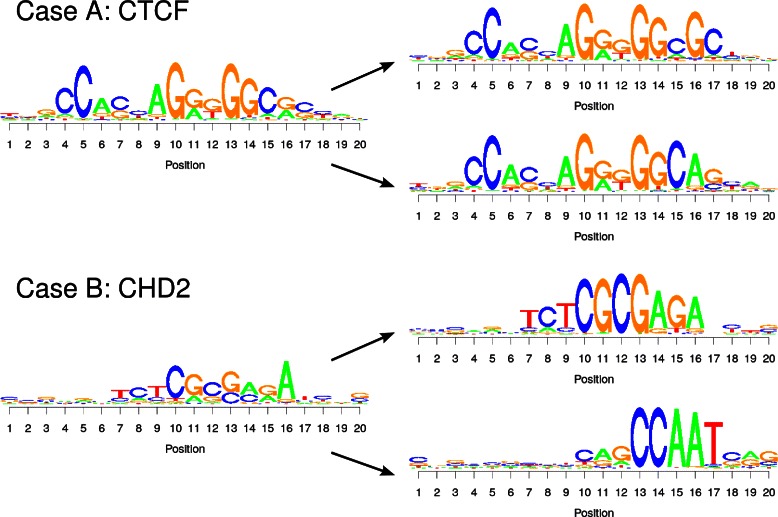
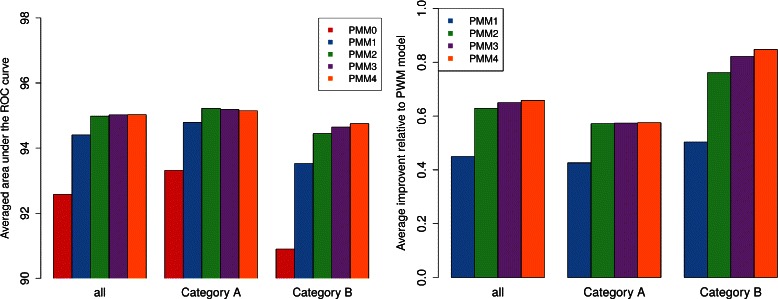
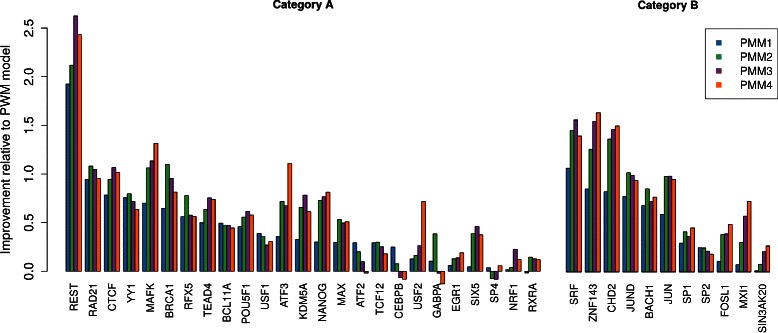
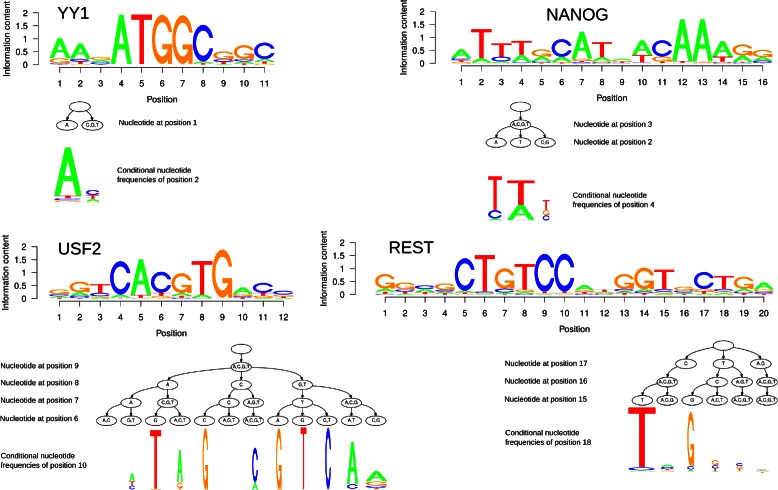
Results: To address this issue, we propose a stochastic algorithm for performing robust model selection in a latent variable setting. This algorithm yields a solution without relying on hyperparameter-tuning via massive cross-validation or other computationally expensive resampling techniques. Using this algorithm for learning inhomogeneous parsimonious Markov models, we study the degree of putative higher-order intra-motif dependencies for transcription factor binding sites inferred via de novo motif discovery from ChIP-seq data. We find that intra-motif dependencies are prevalent and not limited to first-order dependencies among directly adjacent nucleotides, but that second-order models appear to be the significantly better choice.
Conclusions: The traditional PWM model appears to be indeed insufficient to infer realistic sequence motifs, as it is on average outperformed by more complex models that take into account intra-motif dependencies. Moreover, using such models together with an appropriate model selection procedure does not lead to a significant performance loss in comparison with the PWM model for any of the studied transcription factors. Hence, we find it worthwhile to recommend that any modern motif discovery algorithm should attempt to take into account intra-motif dependencies.
Figures
Similar articles
-
Combining phylogenetic footprinting with motif models incorporating intra-motif dependencies.BMC Bioinformatics. 2017 Mar 1;18(1):141. doi: 10.1186/s12859-017-1495-1. BMC Bioinformatics. 2017. PMID: 28249564 Free PMC article.
-
On the value of intra-motif dependencies of human insulator protein CTCF.PLoS One. 2014 Jan 22;9(1):e85629. doi: 10.1371/journal.pone.0085629. eCollection 2014. PLoS One. 2014. PMID: 24465627 Free PMC article.
-
Bayesian Markov models consistently outperform PWMs at predicting motifs in nucleotide sequences.Nucleic Acids Res. 2016 Jul 27;44(13):6055-69. doi: 10.1093/nar/gkw521. Epub 2016 Jun 9. Nucleic Acids Res. 2016. PMID: 27288444 Free PMC article.
-
An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data.Brief Bioinform. 2018 Sep 28;19(5):1069-1081. doi: 10.1093/bib/bbx026. Brief Bioinform. 2018. PMID: 28334268 Review.
-
A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data.Biol Direct. 2014 Feb 20;9:4. doi: 10.1186/1745-6150-9-4. Biol Direct. 2014. PMID: 24555784 Free PMC article. Review.
Cited by
-
A map of direct TF-DNA interactions in the human genome.Nucleic Acids Res. 2019 Feb 28;47(4):e21. doi: 10.1093/nar/gky1210. Nucleic Acids Res. 2019. PMID: 30517703 Free PMC article.
-
Modeling in-vivo protein-DNA binding by combining multiple-instance learning with a hybrid deep neural network.Sci Rep. 2019 Jun 11;9(1):8484. doi: 10.1038/s41598-019-44966-x. Sci Rep. 2019. PMID: 31186519 Free PMC article.
-
The orientation of transcription factor binding site motifs in gene promoter regions: does it matter?BMC Genomics. 2016 Mar 3;17:185. doi: 10.1186/s12864-016-2549-x. BMC Genomics. 2016. PMID: 26939991 Free PMC article.
-
CircularLogo: A lightweight web application to visualize intra-motif dependencies.BMC Bioinformatics. 2017 May 22;18(1):269. doi: 10.1186/s12859-017-1680-2. BMC Bioinformatics. 2017. PMID: 28532394 Free PMC article.
-
InMoDe: tools for learning and visualizing intra-motif dependencies of DNA binding sites.Bioinformatics. 2017 Feb 15;33(4):580-582. doi: 10.1093/bioinformatics/btw689. Bioinformatics. 2017. PMID: 28035026 Free PMC article.
References
-
- Zhang MQ, Marr TG. A weight array method for splicing signals analysis. Comput Appl Biosci. 1993;9:499–509. - PubMed
-
- Barash Y, Elidan G, Friedman N, Kaplan T. Proceedings of the Seventh Annual International Conference on Research in Computational Molecular Biology. NY, USA: ACM; 2003. Modeling dependencies in protein-DNA binding sites.
-
- Rahmann S, Müller T, Vingron M. On the power of profiles for transcription factor binding site detection. Stat Appl Genet Molec Biol. 2003;2(1):1544–6115. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
