

Amphetamine activates Rho GTPase signaling to mediate dopamine transporter internalization and acute behavioral effects of amphetamine
- PMID: 26553986
- PMCID: PMC4697400
- DOI: 10.1073/pnas.1511670112
Amphetamine activates Rho GTPase signaling to mediate dopamine transporter internalization and acute behavioral effects of amphetamine
Abstract
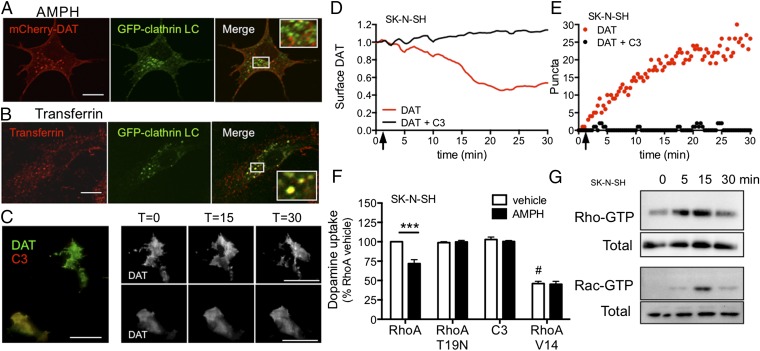
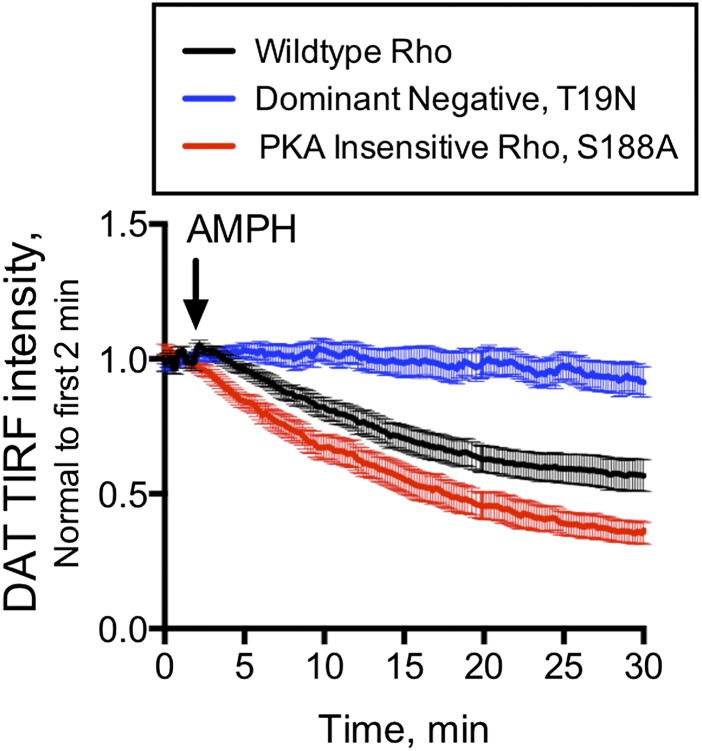
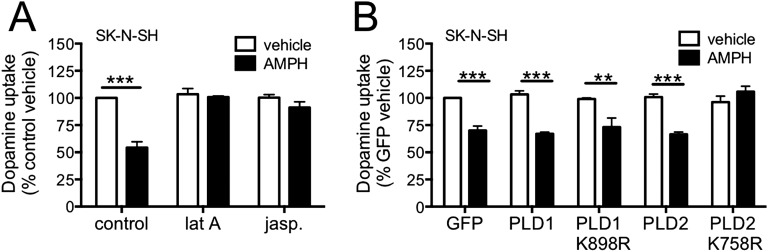
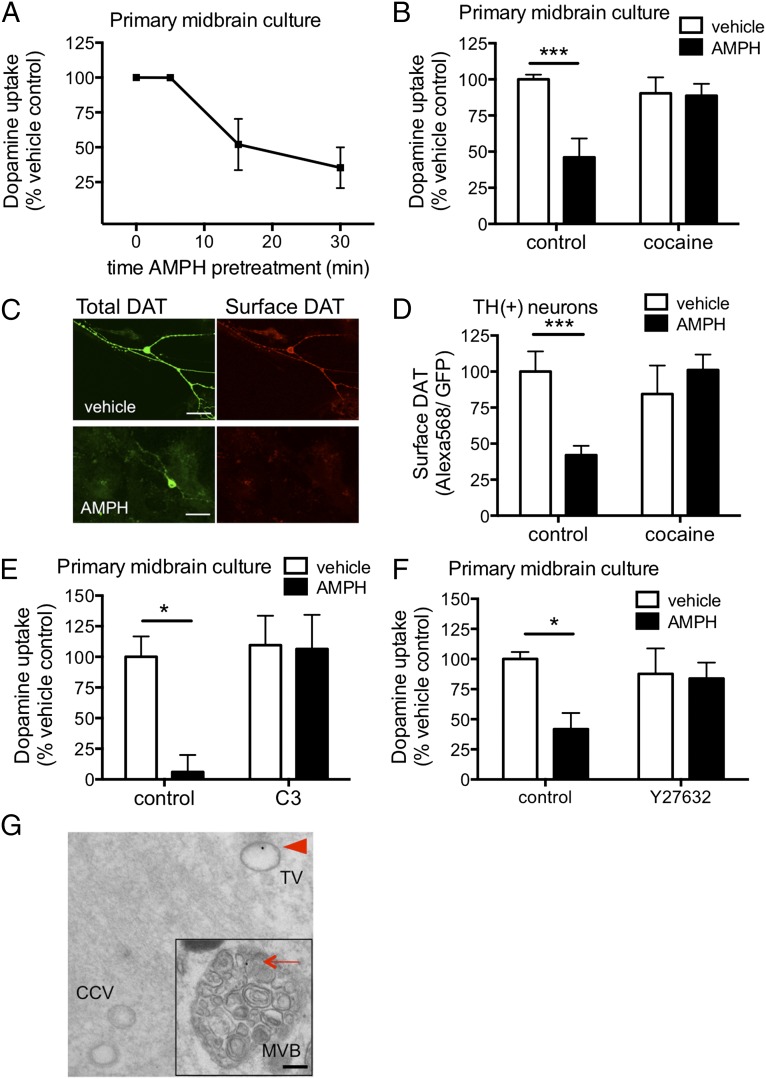
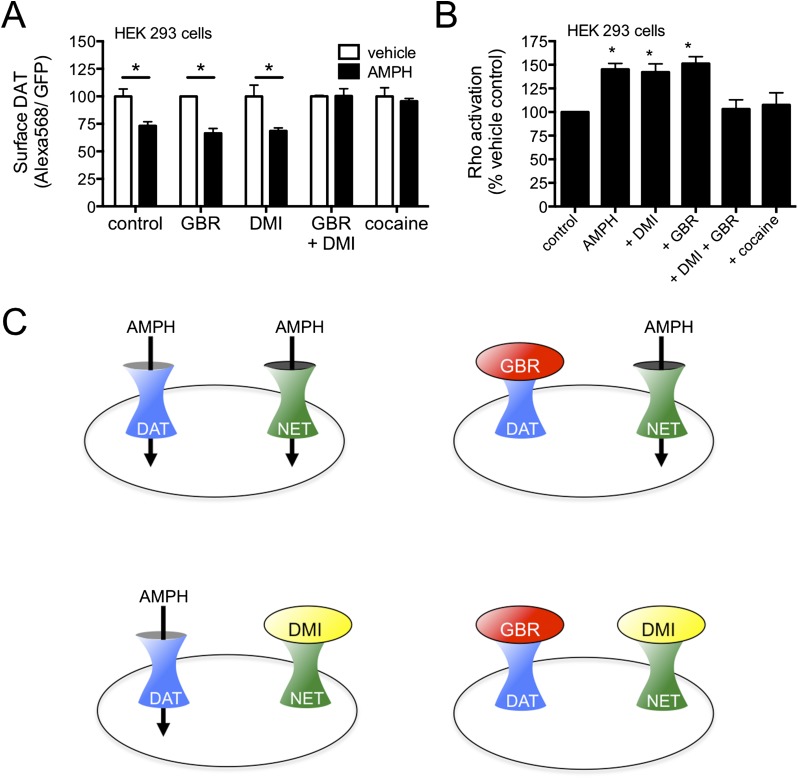
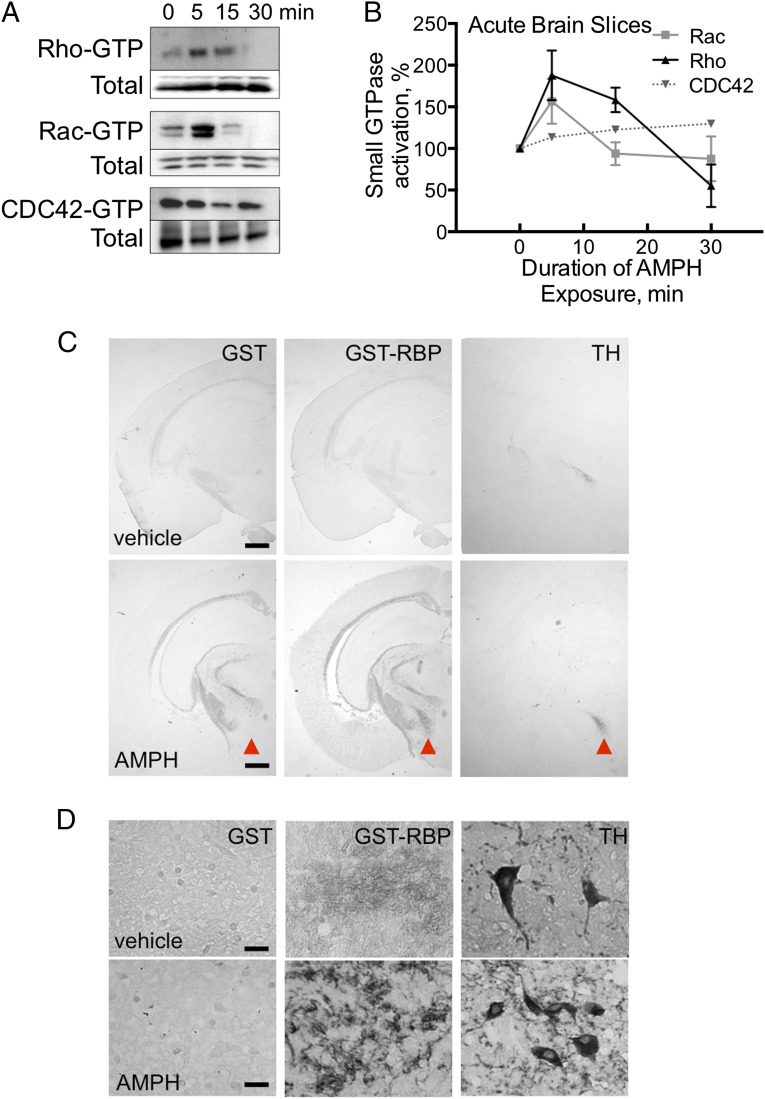
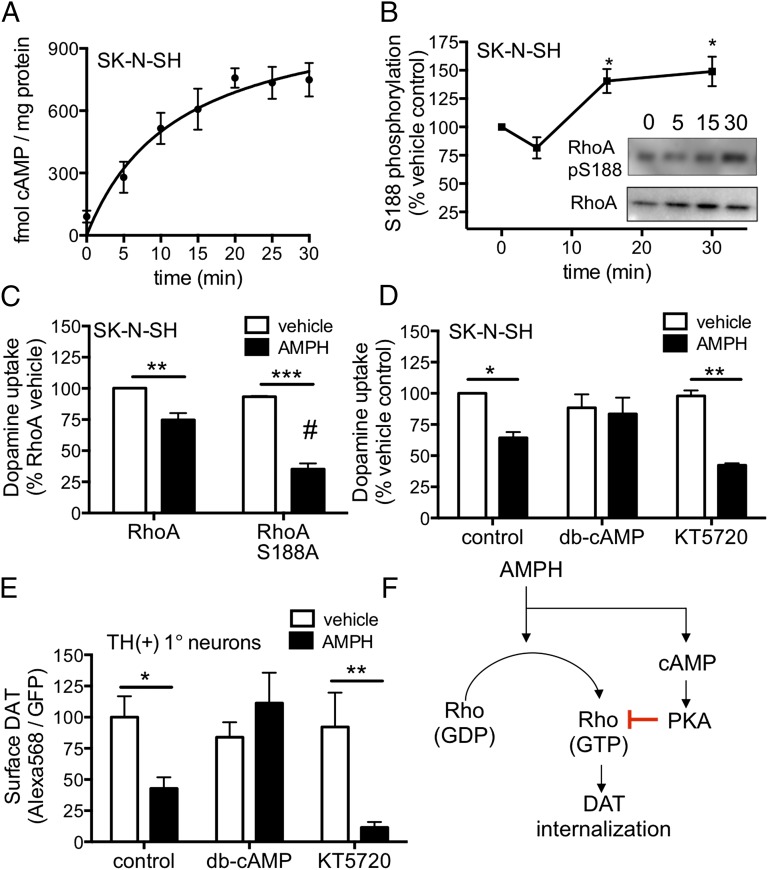
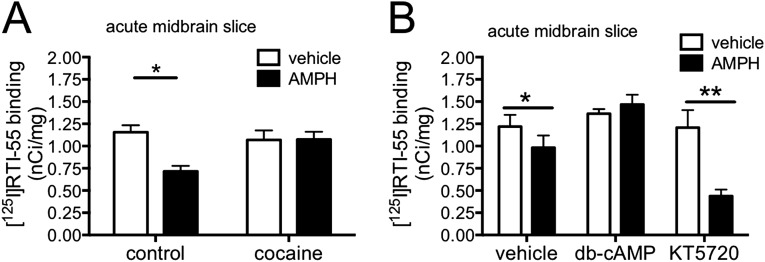
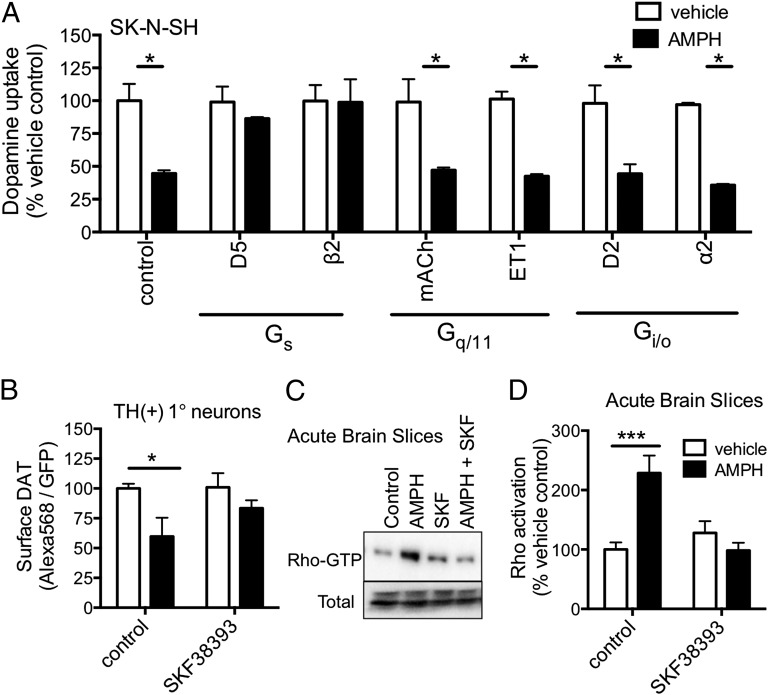
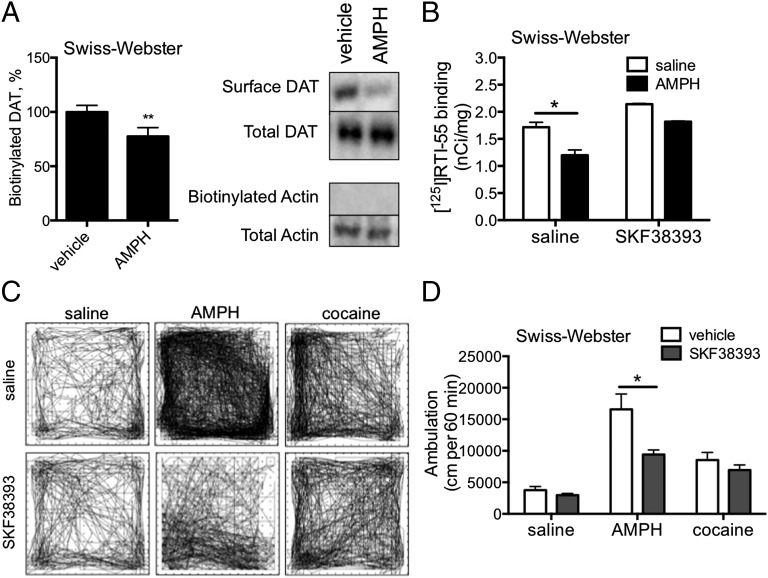
Acute amphetamine (AMPH) exposure elevates extracellular dopamine through a variety of mechanisms that include inhibition of dopamine reuptake, depletion of vesicular stores, and facilitation of dopamine efflux across the plasma membrane. Recent work has shown that the DAT substrate AMPH, unlike cocaine and other nontransported blockers, can also stimulate endocytosis of the plasma membrane dopamine transporter (DAT). Here, we show that when AMPH enters the cytoplasm it rapidly stimulates DAT internalization through a dynamin-dependent, clathrin-independent process. This effect, which can be observed in transfected cells, cultured dopamine neurons, and midbrain slices, is mediated by activation of the small GTPase RhoA. Inhibition of RhoA activity with C3 exotoxin or a dominant-negative RhoA blocks AMPH-induced DAT internalization. These actions depend on AMPH entry into the cell and are blocked by the DAT inhibitor cocaine. AMPH also stimulates cAMP accumulation and PKA-dependent inactivation of RhoA, thus providing a mechanism whereby PKA- and RhoA-dependent signaling pathways can interact to regulate the timing and robustness of AMPH's effects on DAT internalization. Consistent with this model, the activation of D1/D5 receptors that couple to PKA in dopamine neurons antagonizes RhoA activation, DAT internalization, and hyperlocomotion observed in mice after AMPH treatment. These observations support the existence of an unanticipated intracellular target that mediates the effects of AMPH on RhoA and cAMP signaling and suggest new pathways to target to disrupt AMPH action.
Keywords: Rho GTPase; amphetamine; dopamine transporter; endocytosis; protein kinase A.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Insights in how amphetamine ROCKs (Rho-associated containing kinase) membrane protein trafficking.Proc Natl Acad Sci U S A. 2015 Dec 22;112(51):15538-9. doi: 10.1073/pnas.1520960112. Epub 2015 Nov 25. Proc Natl Acad Sci U S A. 2015. PMID: 26607447 Free PMC article. No abstract available.
Similar articles
-
G protein-coupled receptor signaling in VTA dopaminergic neurons bidirectionally regulates the acute locomotor response to amphetamine but does not affect behavioral sensitization.Neuropharmacology. 2019 Dec 15;161:107663. doi: 10.1016/j.neuropharm.2019.06.002. Epub 2019 Jun 4. Neuropharmacology. 2019. PMID: 31173760
-
Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism.Proc Natl Acad Sci U S A. 2000 Jun 6;97(12):6850-5. doi: 10.1073/pnas.110035297. Proc Natl Acad Sci U S A. 2000. PMID: 10823899 Free PMC article.
-
Altered baseline and amphetamine-mediated behavioral profiles in dopamine transporter Cre (DAT-Ires-Cre) mice compared to tyrosine hydroxylase Cre (TH-Cre) mice.Psychopharmacology (Berl). 2020 Dec;237(12):3553-3568. doi: 10.1007/s00213-020-05635-4. Epub 2020 Aug 10. Psychopharmacology (Berl). 2020. PMID: 32778904 Free PMC article.
-
Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter.J Neurosci. 1998 Mar 15;18(6):1979-86. doi: 10.1523/JNEUROSCI.18-06-01979.1998. J Neurosci. 1998. PMID: 9482784 Free PMC article. Review.
-
Phosphorylation of the Amino Terminus of the Dopamine Transporter: Regulatory Mechanisms and Implications for Amphetamine Action.Adv Pharmacol. 2018;82:205-234. doi: 10.1016/bs.apha.2017.09.002. Epub 2017 Oct 25. Adv Pharmacol. 2018. PMID: 29413521 Free PMC article. Review.
Cited by
-
Regulation of Glutamate, GABA and Dopamine Transporter Uptake, Surface Mobility and Expression.Front Cell Neurosci. 2021 Apr 13;15:670346. doi: 10.3389/fncel.2021.670346. eCollection 2021. Front Cell Neurosci. 2021. PMID: 33927596 Free PMC article. Review.
-
Trace amine-associated receptor 1 and drug abuse.Adv Pharmacol. 2022;93:373-401. doi: 10.1016/bs.apha.2021.10.005. Epub 2021 Nov 11. Adv Pharmacol. 2022. PMID: 35341572 Free PMC article.
-
The Use of Drosophila to Understand Psychostimulant Responses.Biomedicines. 2022 Jan 6;10(1):119. doi: 10.3390/biomedicines10010119. Biomedicines. 2022. PMID: 35052798 Free PMC article. Review.
-
Heterogeneities in Axonal Structure and Transporter Distribution Lower Dopamine Reuptake Efficiency.eNeuro. 2018 Feb 5;5(1):ENEURO.0298-17.2017. doi: 10.1523/ENEURO.0298-17.2017. eCollection 2018 Jan-Feb. eNeuro. 2018. PMID: 29430519 Free PMC article.
-
Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption.Brain Sci. 2019 Jun 30;9(7):155. doi: 10.3390/brainsci9070155. Brain Sci. 2019. PMID: 31262025 Free PMC article.
References
-
- Gonzalez Castro F, Barrington EH, Walton MA, Rawson RA. Cocaine and methamphetamine: Differential addiction rates. Psychol Addict Behav. 2000;14(4):390–396. - PubMed
-
- Kalechstein AD, et al. Psychiatric comorbidity of methamphetamine dependence in a forensic sample. J Neuropsychiatry Clin Neurosci. 2000;12(4):480–484. - PubMed
-
- Rawson R, et al. Methamphetamine and cocaine users: Differences in characteristics and treatment retention. J Psychoactive Drugs. 2000;32(2):233–238. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
