

HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart
- PMID: 26553996
- PMCID: PMC4664353
- DOI: 10.1073/pnas.1508760112
HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart
Abstract
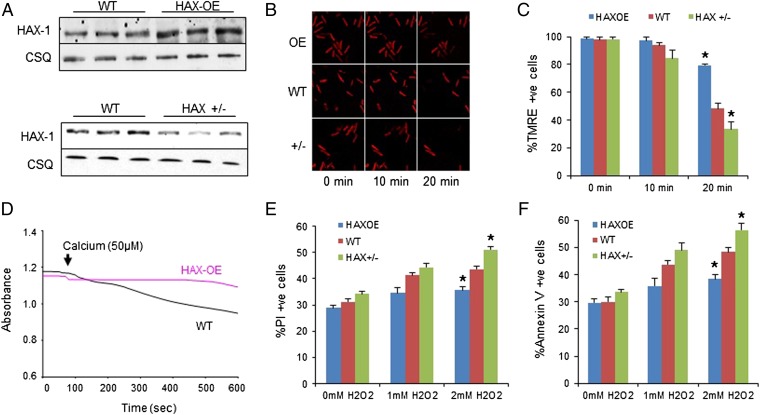
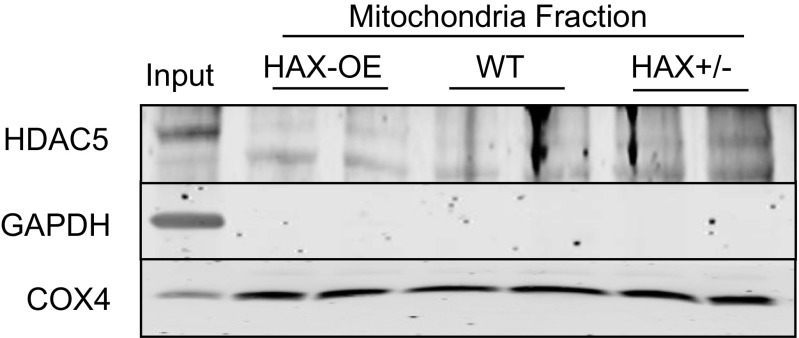
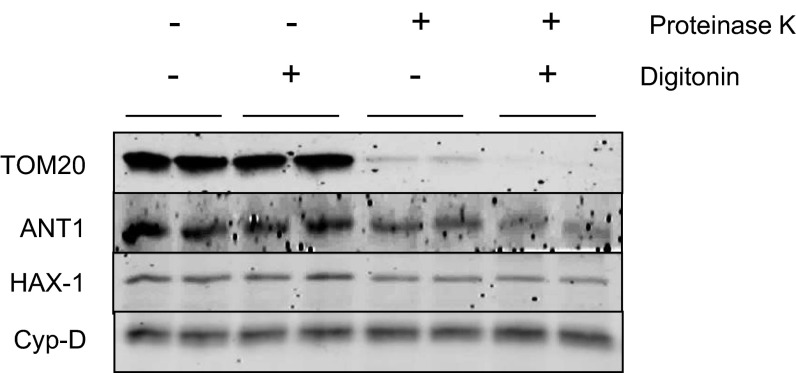
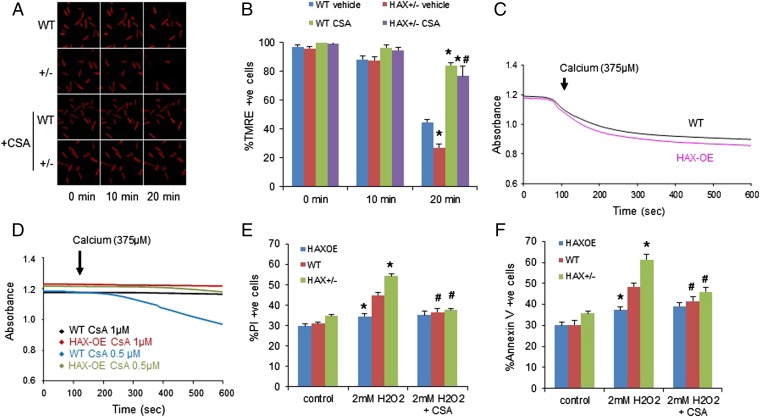
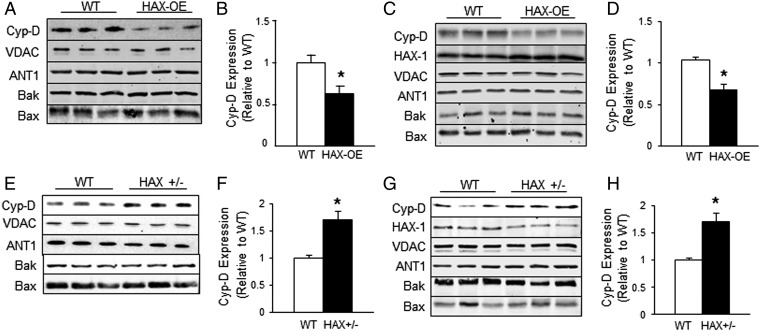
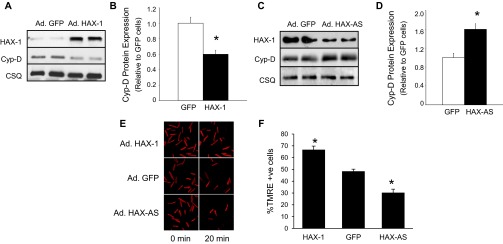
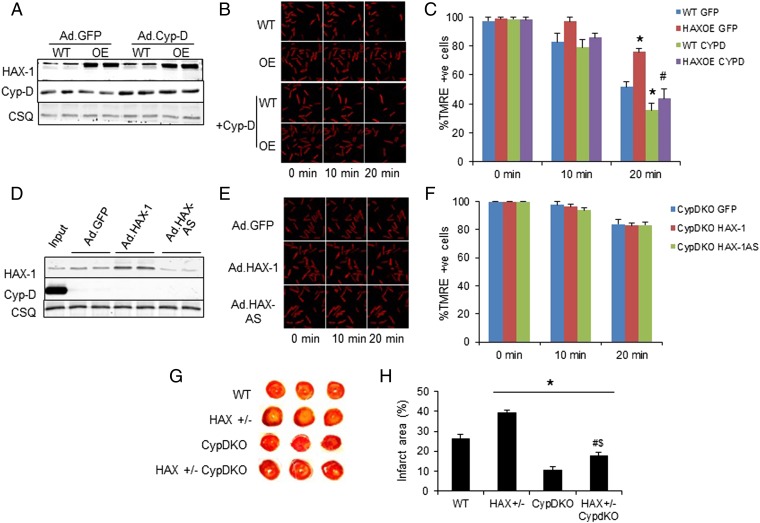
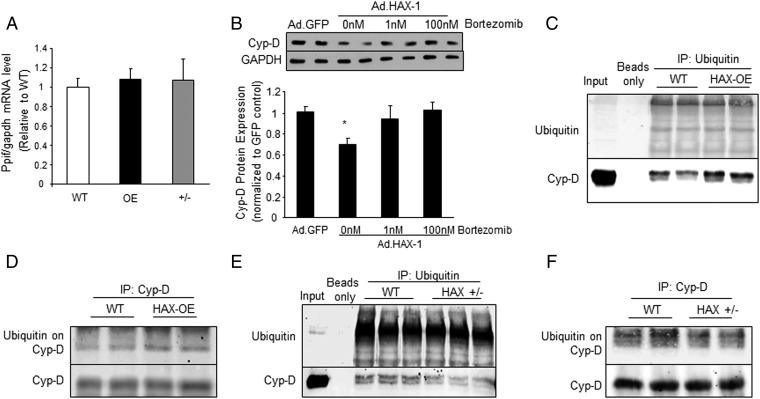
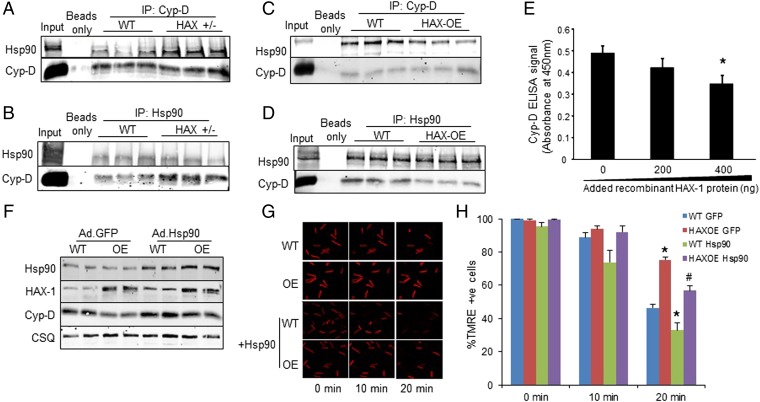
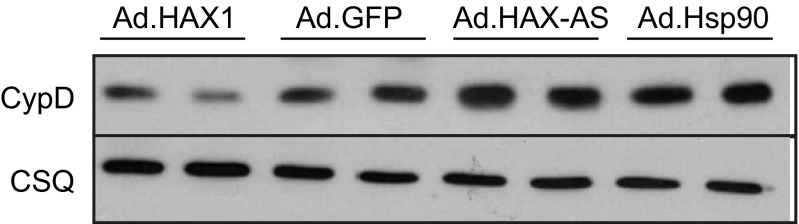
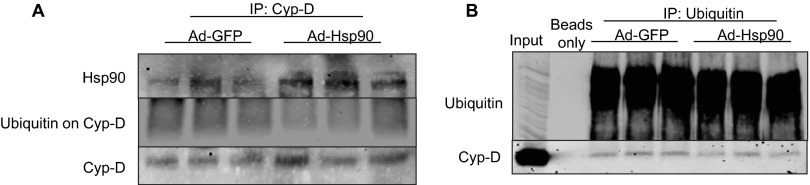
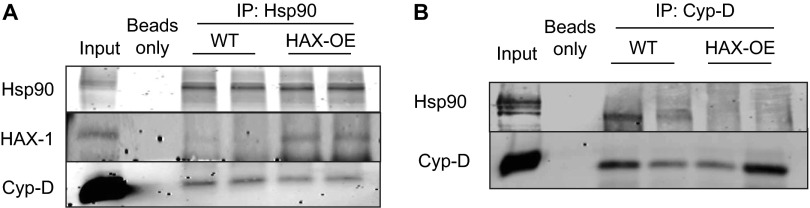
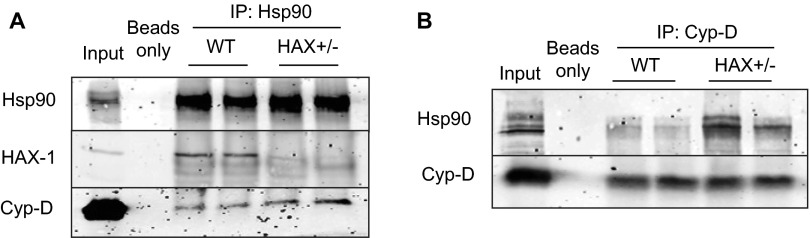
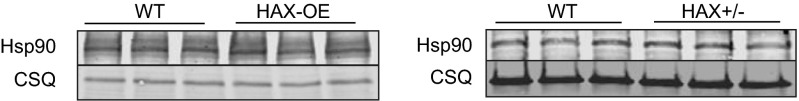
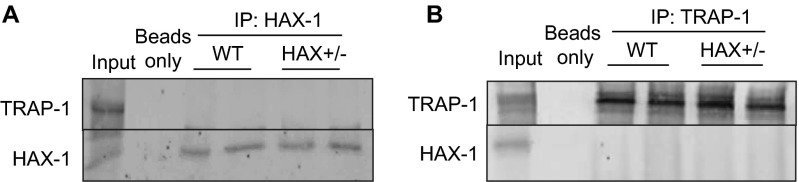
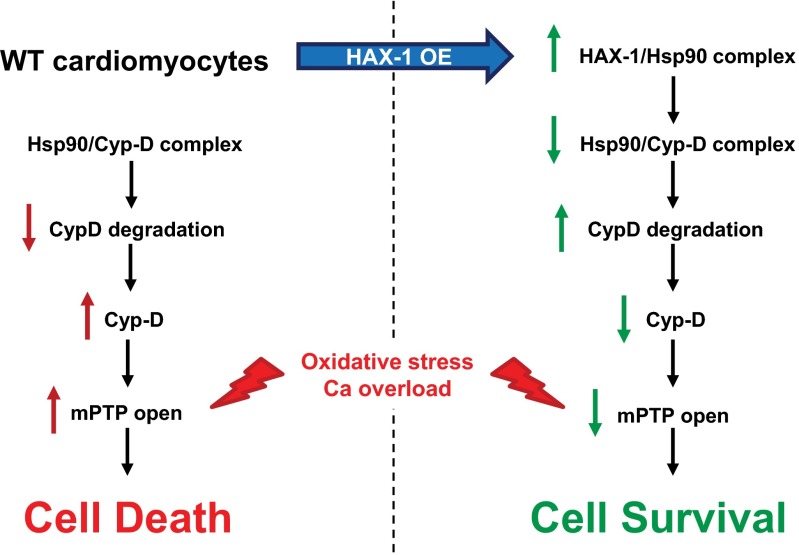
The major underpinning of massive cell death associated with myocardial infarction involves opening of the mitochondrial permeability transition pore (mPTP), resulting in disruption of mitochondria membrane integrity and programmed necrosis. Studies in human lymphocytes suggested that the hematopoietic-substrate-1 associated protein X-1 (HAX-1) is linked to regulation of mitochondrial membrane function, but its role in controlling mPTP activity remains obscure. Herein we used models with altered HAX-1 expression levels in the heart and uncovered an unexpected role of HAX-1 in regulation of mPTP and cardiomyocyte survival. Cardiac-specific HAX-1 overexpression was associated with resistance against loss of mitochondrial membrane potential, induced by oxidative stress, whereas HAX-1 heterozygous deficiency exacerbated vulnerability. The protective effects of HAX-1 were attributed to specific down-regulation of cyclophilin-D levels leading to reduction in mPTP activation. Accordingly, cyclophilin-D and mPTP were increased in heterozygous hearts, but genetic ablation of cyclophilin-D in these hearts significantly alleviated their susceptibility to ischemia/reperfusion injury. Mechanistically, alterations in cyclophilin-D levels by HAX-1 were contributed by the ubiquitin-proteosomal degradation pathway. HAX-1 overexpression enhanced cyclophilin-D ubiquitination, whereas proteosomal inhibition restored cyclophilin-D levels. The regulatory effects of HAX-1 were mediated through interference of cyclophilin-D binding to heat shock protein-90 (Hsp90) in mitochondria, rendering it susceptible to degradation. Accordingly, enhanced Hsp90 expression in HAX-1 overexpressing cardiomyocytes increased cyclophilin-D levels, as well as mPTP activation upon oxidative stress. Taken together, our findings reveal the role of HAX-1 in regulating cyclophilin-D levels via an Hsp90-dependent mechanism, resulting in protection against activation of mPTP and subsequent cell death responses.
Keywords: HAX-1; cyclophilin-D; heat shock protein-90; mitochondrial permeability transition; necrosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
HAX-1 regulates SERCA2a oxidation and degradation.J Mol Cell Cardiol. 2018 Jan;114:220-233. doi: 10.1016/j.yjmcc.2017.11.014. Epub 2017 Nov 21. J Mol Cell Cardiol. 2018. PMID: 29169992 Free PMC article.
-
Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90.Circ Res. 2013 Jan 4;112(1):79-89. doi: 10.1161/CIRCRESAHA.112.279935. Epub 2012 Sep 14. Circ Res. 2013. PMID: 22982986 Free PMC article.
-
Increased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophy.J Mol Cell Cardiol. 2009 Mar;46(3):420-30. doi: 10.1016/j.yjmcc.2008.10.020. Epub 2008 Nov 6. J Mol Cell Cardiol. 2009. PMID: 19094991
-
Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore.Circ J. 2013;77(5):1111-22. doi: 10.1253/circj.cj-13-0321. Epub 2013 Mar 29. Circ J. 2013. PMID: 23538482 Free PMC article. Review.
-
Mitochondrial permeability transition in cardiac ischemia-reperfusion: whether cyclophilin D is a viable target for cardioprotection?Cell Mol Life Sci. 2017 Aug;74(15):2795-2813. doi: 10.1007/s00018-017-2502-4. Epub 2017 Apr 4. Cell Mol Life Sci. 2017. PMID: 28378042 Free PMC article. Review.
Cited by
-
Role of Oxidative Stress in Reperfusion following Myocardial Ischemia and Its Treatments.Oxid Med Cell Longev. 2021 May 18;2021:6614009. doi: 10.1155/2021/6614009. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34055195 Free PMC article. Review.
-
Ablation of phospholamban rescues reperfusion arrhythmias but exacerbates myocardium infarction in hearts with Ca2+/calmodulin kinase II constitutive phosphorylation of ryanodine receptors.Cardiovasc Res. 2019 Mar 1;115(3):556-569. doi: 10.1093/cvr/cvy213. Cardiovasc Res. 2019. PMID: 30169578 Free PMC article.
-
Furanoditerpenes from Spongia (Spongia) tubulifera Display Mitochondrial-Mediated Neuroprotective Effects by Targeting Cyclophilin D.ACS Chem Neurosci. 2022 Aug 17;13(16):2449-2463. doi: 10.1021/acschemneuro.2c00208. Epub 2022 Jul 28. ACS Chem Neurosci. 2022. PMID: 35901231 Free PMC article.
-
HAX-1 regulates SERCA2a oxidation and degradation.J Mol Cell Cardiol. 2018 Jan;114:220-233. doi: 10.1016/j.yjmcc.2017.11.014. Epub 2017 Nov 21. J Mol Cell Cardiol. 2018. PMID: 29169992 Free PMC article.
-
Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis?Cell Mol Life Sci. 2016 Jun;73(11-12):2309-24. doi: 10.1007/s00018-016-2202-5. Epub 2016 Apr 5. Cell Mol Life Sci. 2016. PMID: 27048819 Free PMC article. Review.
References
-
- Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. 2012;111(9):1198–1207. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
