

Designed protein reveals structural determinants of extreme kinetic stability
- PMID: 26554002
- PMCID: PMC4664365
- DOI: 10.1073/pnas.1510748112
Designed protein reveals structural determinants of extreme kinetic stability
Abstract
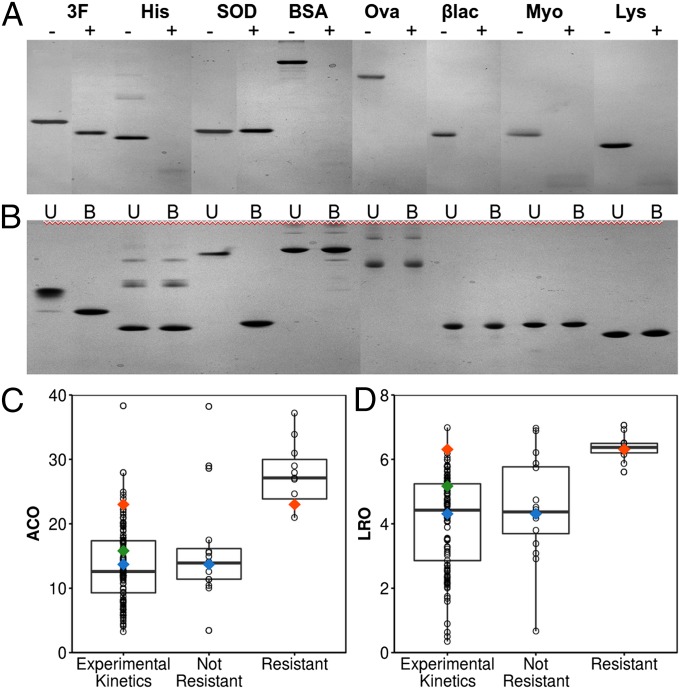
The design of stable, functional proteins is difficult. Improved design requires a deeper knowledge of the molecular basis for design outcomes and properties. We previously used a bioinformatics and energy function method to design a symmetric superfold protein composed of repeating structural elements with multivalent carbohydrate-binding function, called ThreeFoil. This and similar methods have produced a notably high yield of stable proteins. Using a battery of experimental and computational analyses we show that despite its small size and lack of disulfide bonds, ThreeFoil has remarkably high kinetic stability and its folding is specifically chaperoned by carbohydrate binding. It is also extremely stable against thermal and chemical denaturation and proteolytic degradation. We demonstrate that the kinetic stability can be predicted and modeled using absolute contact order (ACO) and long-range order (LRO), as well as coarse-grained simulations; the stability arises from a topology that includes many long-range contacts which create a large and highly cooperative energy barrier for unfolding and folding. Extensive data from proteomic screens and other experiments reveal that a high ACO/LRO is a general feature of proteins with strong resistances to denaturation and degradation. These results provide tractable approaches for predicting resistance and designing proteins with sufficient topological complexity and long-range interactions to accommodate destabilizing functional features as well as withstand chemical and proteolytic challenge.
Keywords: SDS/protease resistance; coarse-grained simulations; contact order; protein folding; protein topology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Engineering the kinetic stability of a β-trefoil protein by tuning its topological complexity.Front Mol Biosci. 2023 Feb 8;10:1021733. doi: 10.3389/fmolb.2023.1021733. eCollection 2023. Front Mol Biosci. 2023. PMID: 36845544 Free PMC article.
-
Protein unfolding rates correlate as strongly as folding rates with native structure.Protein Sci. 2015 Apr;24(4):580-7. doi: 10.1002/pro.2606. Epub 2014 Dec 26. Protein Sci. 2015. PMID: 25422093 Free PMC article.
-
Designing cooperativity into the designed protein Top7.Proteins. 2014 Mar;82(3):364-74. doi: 10.1002/prot.24393. Epub 2013 Oct 17. Proteins. 2014. PMID: 23966061
-
Local versus nonlocal interactions in protein folding and stability--an experimentalist's point of view.Fold Des. 1996;1(4):R71-7. doi: 10.1016/S1359-0278(96)00036-3. Fold Des. 1996. PMID: 9079385 Review.
-
Cooperativity, local-nonlocal coupling, and nonnative interactions: principles of protein folding from coarse-grained models.Annu Rev Phys Chem. 2011;62:301-26. doi: 10.1146/annurev-physchem-032210-103405. Annu Rev Phys Chem. 2011. PMID: 21453060 Review.
Cited by
-
Computational design of symmetrical eight-bladed β-propeller proteins.IUCrJ. 2019 Jan 1;6(Pt 1):46-55. doi: 10.1107/S205225251801480X. eCollection 2019 Jan 1. IUCrJ. 2019. PMID: 30713702 Free PMC article.
-
Computational tools help improve protein stability but with a solubility tradeoff.J Biol Chem. 2017 Sep 1;292(35):14349-14361. doi: 10.1074/jbc.M117.784165. Epub 2017 Jul 14. J Biol Chem. 2017. PMID: 28710274 Free PMC article.
-
An extensively glycosylated archaeal pilus survives extreme conditions.Nat Microbiol. 2019 Aug;4(8):1401-1410. doi: 10.1038/s41564-019-0458-x. Epub 2019 May 20. Nat Microbiol. 2019. PMID: 31110358 Free PMC article.
-
Cooperative hydrophobic core interactions in the β-trefoil architecture.Protein Sci. 2021 May;30(5):956-965. doi: 10.1002/pro.4059. Epub 2021 Mar 16. Protein Sci. 2021. PMID: 33686691 Free PMC article.
-
Engineering the kinetic stability of a β-trefoil protein by tuning its topological complexity.Front Mol Biosci. 2023 Feb 8;10:1021733. doi: 10.3389/fmolb.2023.1021733. eCollection 2023. Front Mol Biosci. 2023. PMID: 36845544 Free PMC article.
References
-
- Broom A, et al. Modular evolution and the origins of symmetry: Reconstruction of a three-fold symmetric globular protein. Structure. 2012;20(1):161–171. - PubMed
-
- Longo LM, Kumru OS, Middaugh CR, Blaber M. Evolution and design of protein structure by folding nucleus symmetric expansion. Structure. 2014;22(10):1377–1384. - PubMed
-
- Thomson AR, et al. Computational design of water-soluble α-helical barrels. Science. 2014;346(6208):485–488. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
