

Pathophysiological mechanisms of death resistance in colorectal carcinoma
- PMID: 26557002
- PMCID: PMC4631976
- DOI: 10.3748/wjg.v21.i41.11777
Pathophysiological mechanisms of death resistance in colorectal carcinoma
Abstract
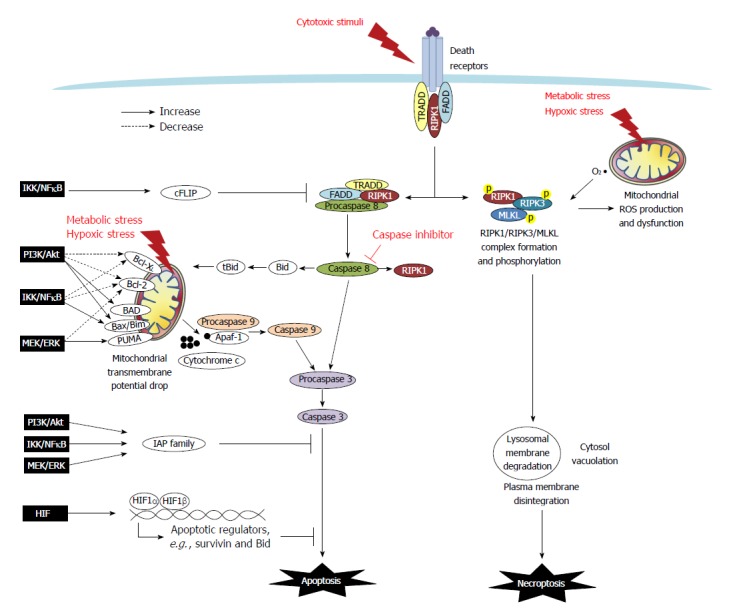
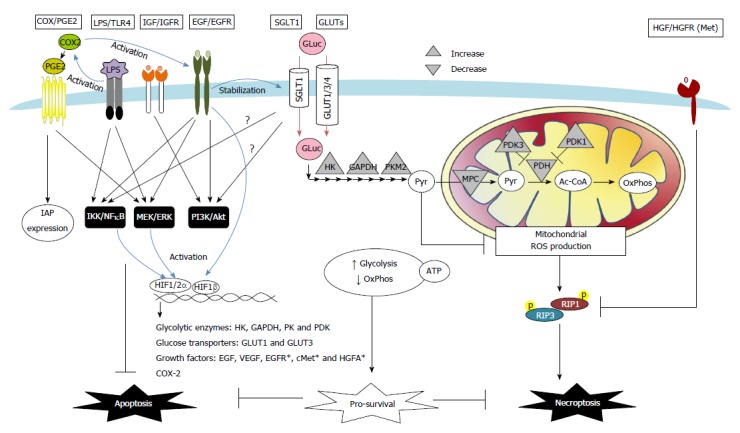
Colon cancers develop adaptive mechanisms to survive under extreme conditions and display hallmarks of unlimited proliferation and resistance to cell death. The deregulation of cell death is a key factor that contributes to chemoresistance in tumors. In a physiological context, balance between cell proliferation and death, and protection against cell damage are fundamental processes for maintaining gut epithelial homeostasis. The mechanisms underlying anti-death cytoprotection and tumor resistance often bear common pathways, and although distinguishing them would be a challenge, it would also provide an opportunity to develop advanced anti-cancer therapeutics. This review will outline cell death pathways (i.e., apoptosis, necrosis, and necroptosis), and discuss cytoprotective strategies in normal intestinal epithelium and death resistance mechanisms of colon tumor. In colorectal cancers, the intracellular mechanisms of death resistance include the direct alteration of apoptotic and necroptotic machinery and the upstream events modulating death effectors such as tumor suppressor gene inactivation and pro-survival signaling pathways. The autocrine, paracrine and exogenous factors within a tumor microenvironment can also instigate resistance against apoptotic and necroptotic cell death in colon cancers through changes in receptor signaling or transporter uptake. The roles of cyclooxygenase-2/prostaglandin E2, growth factors, glucose, and bacterial lipopolysaccharides in colorectal cancer will be highlighted. Targeting anti-death pathways in the colon cancer tissue might be a promising approach outside of anti-proliferation and anti-angiogenesis strategies for developing novel drugs to treat refractory tumors.
Keywords: Anti-apoptosis; Anti-necroptosis; Chemoresistance; Colon cancer; Cytoprotection; Tumorigenesis.
Figures
Similar articles
-
Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: paving the road to patient-tailored therapy.Drug Resist Updat. 2004 Dec;7(6):345-58. doi: 10.1016/j.drup.2004.11.002. Epub 2005 Jan 8. Drug Resist Updat. 2004. PMID: 15790545 Review.
-
Apoptosis and colorectal cancer: implications for therapy.Trends Mol Med. 2009 May;15(5):225-33. doi: 10.1016/j.molmed.2009.03.003. Epub 2009 Apr 8. Trends Mol Med. 2009. PMID: 19362056 Review.
-
Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach.Drug Resist Updat. 2015 Nov;23:69-78. doi: 10.1016/j.drup.2015.08.004. Epub 2015 Aug 22. Drug Resist Updat. 2015. PMID: 26341193 Review.
-
Isoledene from Mesua ferrea oleo-gum resin induces apoptosis in HCT 116 cells through ROS-mediated modulation of multiple proteins in the apoptotic pathways: A mechanistic study.Toxicol Lett. 2016 Aug 22;257:84-96. doi: 10.1016/j.toxlet.2016.05.027. Epub 2016 Jun 3. Toxicol Lett. 2016. PMID: 27268964
-
Apoptosis and chemo-resistance in colorectal cancer.J Surg Oncol. 2007 Jul 1;96(1):77-88. doi: 10.1002/jso.20785. J Surg Oncol. 2007. PMID: 17443738 Review.
Cited by
-
Clinicopathological and Prognostic Value of Necroptosis-Associated lncRNA Model in Patients with Kidney Renal Clear Cell Carcinoma.Dis Markers. 2022 May 23;2022:5204831. doi: 10.1155/2022/5204831. eCollection 2022. Dis Markers. 2022. PMID: 35664432 Free PMC article.
-
The Phenolic compound Kaempferol overcomes 5-fluorouracil resistance in human resistant LS174 colon cancer cells.Sci Rep. 2019 Jan 17;9(1):195. doi: 10.1038/s41598-018-36808-z. Sci Rep. 2019. PMID: 30655588 Free PMC article.
-
miR-365a-3p regulates ADAM10-JAK-STAT signaling to suppress the growth and metastasis of colorectal cancer cells.J Cancer. 2020 Mar 26;11(12):3634-3644. doi: 10.7150/jca.42731. eCollection 2020. J Cancer. 2020. PMID: 32284760 Free PMC article.
-
Glucose Metabolites Exert Opposing Roles in Tumor Chemoresistance.Front Oncol. 2019 Nov 21;9:1282. doi: 10.3389/fonc.2019.01282. eCollection 2019. Front Oncol. 2019. PMID: 31824857 Free PMC article.
-
Apoptotic and Necroptotic Mediators are Differentially Expressed in Mucinous and Non-Mucinous Colorectal Cancer.Front Oncol. 2022 Jul 14;12:815001. doi: 10.3389/fonc.2022.815001. eCollection 2022. Front Oncol. 2022. PMID: 35912268 Free PMC article.
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Cuezva JM, Krajewska M, de Heredia ML, Krajewski S, Santamaría G, Kim H, Zapata JM, Marusawa H, Chamorro M, Reed JC. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 2002;62:6674–6681. - PubMed
-
- Viry E, Paggetti J, Baginska J, Mgrditchian T, Berchem G, Moussay E, Janji B. Autophagy: an adaptive metabolic response to stress shaping the antitumor immunity. Biochem Pharmacol. 2014;92:31–42. - PubMed
-
- Yang SY, Sales KM, Fuller B, Seifalian AM, Winslet MC. Apoptosis and colorectal cancer: implications for therapy. Trends Mol Med. 2009;15:225–233. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
