

Convergence of Hippocampal Pathophysiology in Syngap+/- and Fmr1-/y Mice
- PMID: 26558778
- PMCID: PMC4642239
- DOI: 10.1523/JNEUROSCI.1087-15.2015
Convergence of Hippocampal Pathophysiology in Syngap+/- and Fmr1-/y Mice
Abstract
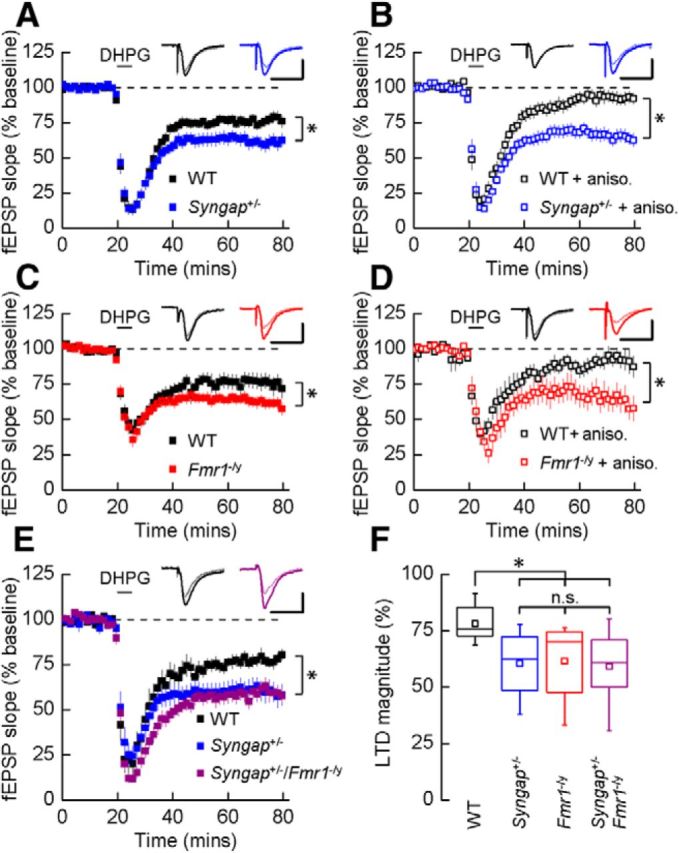
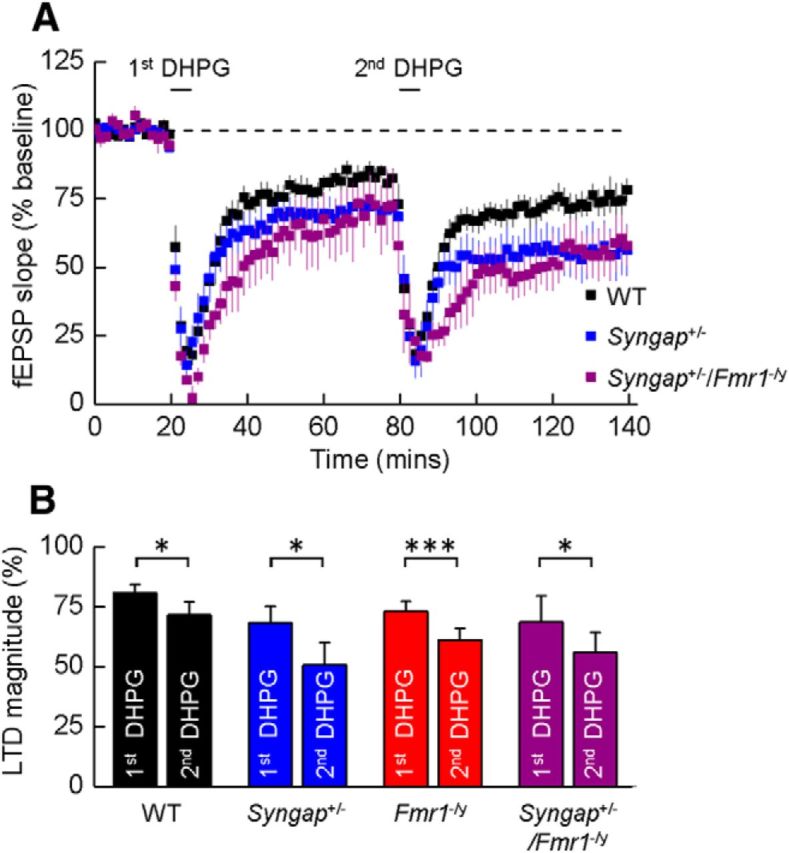
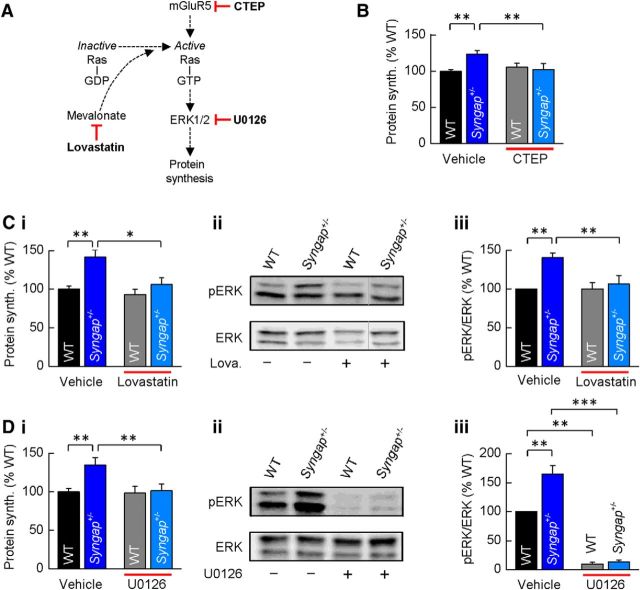
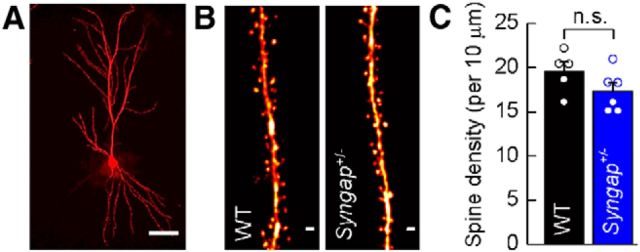
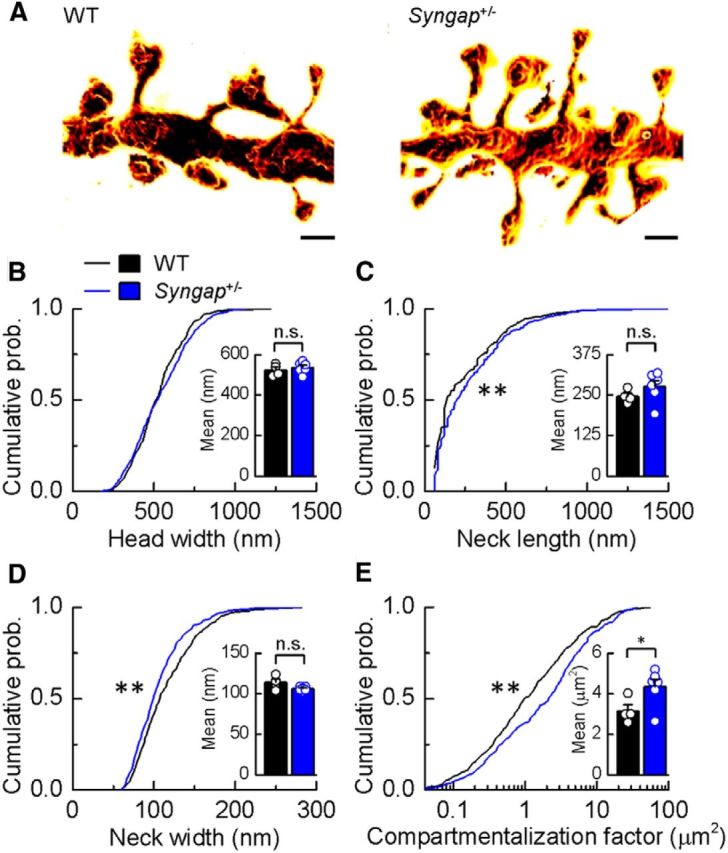
Previous studies have hypothesized that diverse genetic causes of intellectual disability (ID) and autism spectrum disorders (ASDs) converge on common cellular pathways. Testing this hypothesis requires detailed phenotypic analyses of animal models with genetic mutations that accurately reflect those seen in the human condition (i.e., have structural validity) and which produce phenotypes that mirror ID/ASDs (i.e., have face validity). We show that SynGAP haploinsufficiency, which causes ID with co-occurring ASD in humans, mimics and occludes the synaptic pathophysiology associated with deletion of the Fmr1 gene. Syngap(+/-) and Fmr1(-/y) mice show increases in basal protein synthesis and metabotropic glutamate receptor (mGluR)-dependent long-term depression that, unlike in their wild-type controls, is independent of new protein synthesis. Basal levels of phosphorylated ERK1/2 are also elevated in Syngap(+/-) hippocampal slices. Super-resolution microscopy reveals that Syngap(+/-) and Fmr1(-/y) mice show nanoscale alterations in dendritic spine morphology that predict an increase in biochemical compartmentalization. Finally, increased basal protein synthesis is rescued by negative regulators of the mGlu subtype 5 receptor and the Ras-ERK1/2 pathway, indicating that therapeutic interventions for fragile X syndrome may benefit patients with SYNGAP1 haploinsufficiency.
Significance statement: As the genetics of intellectual disability (ID) and autism spectrum disorders (ASDs) are unraveled, a key issue is whether genetically divergent forms of these disorders converge on common biochemical/cellular pathways and hence may be amenable to common therapeutic interventions. This study compares the pathophysiology associated with the loss of fragile X mental retardation protein (FMRP) and haploinsufficiency of synaptic GTPase-activating protein (SynGAP), two prevalent monogenic forms of ID. We show that Syngap(+/-) mice phenocopy Fmr1(-/y) mice in the alterations in mGluR-dependent long-term depression, basal protein synthesis, and dendritic spine morphology. Deficits in basal protein synthesis can be rescued by pharmacological interventions that reduce the mGlu5 receptor-ERK1/2 signaling pathway, which also rescues the same deficit in Fmr1(-/y) mice. Our findings support the hypothesis that phenotypes associated with genetically diverse forms of ID/ASDs result from alterations in common cellular/biochemical pathways.
Keywords: STED; SynGAP; fragile X syndrome; long-term depression; mGluR; neurodevelopmental disorder.
Copyright © 2015 Barnes et al.
Figures

References
-
- Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, Lacaille JC, Niu Z, Eng CM, Yang Y, Palardy S, Belhumeur C, Rouleau GA, Tommerup N, Immken L, Beauchamp MH, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013;34:385–394. doi: 10.1002/humu.22248. - DOI - PubMed
-
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, Xu X, Rumbaugh G. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012;151:709–723. doi: 10.1016/j.cell.2012.08.045. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous