

Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution
- PMID: 26561581
- PMCID: PMC4664355
- DOI: 10.1073/pnas.1519556112
Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution
Erratum in
-
Correction for Ling et al., Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution.Proc Natl Acad Sci U S A. 2016 Feb 2;113(5):E663. doi: 10.1073/pnas.1600151113. Epub 2016 Jan 25. Proc Natl Acad Sci U S A. 2016. PMID: 26811450 Free PMC article. No abstract available.
Abstract
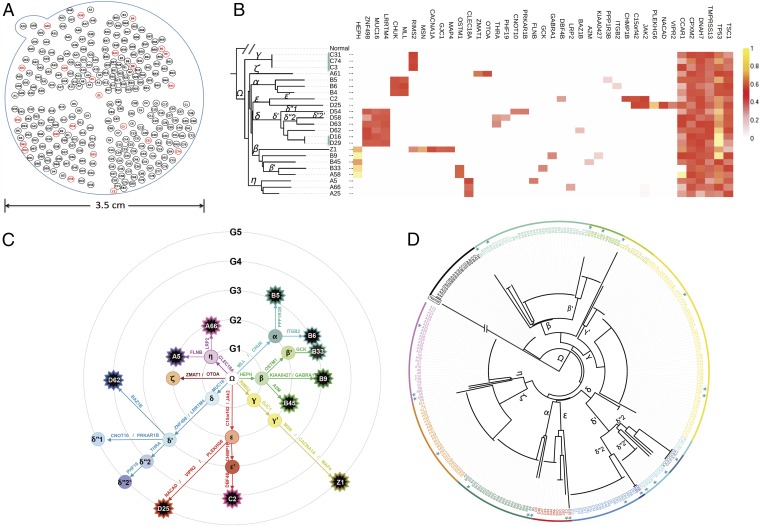
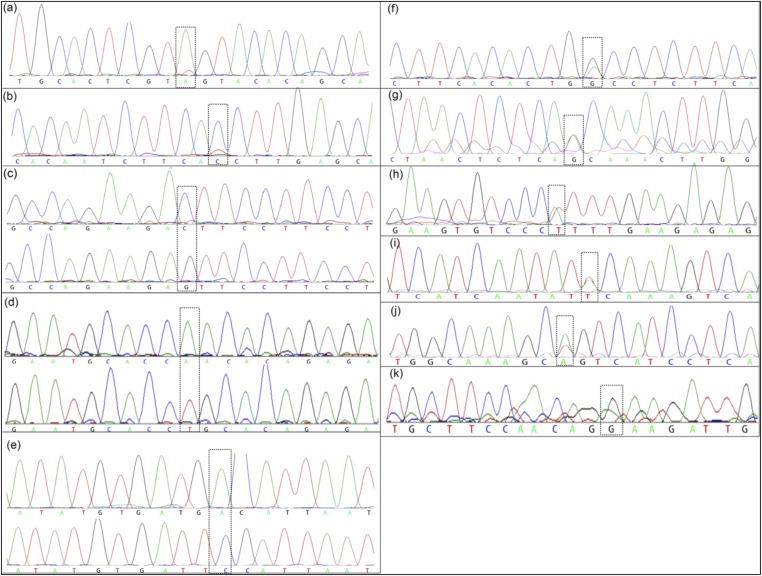
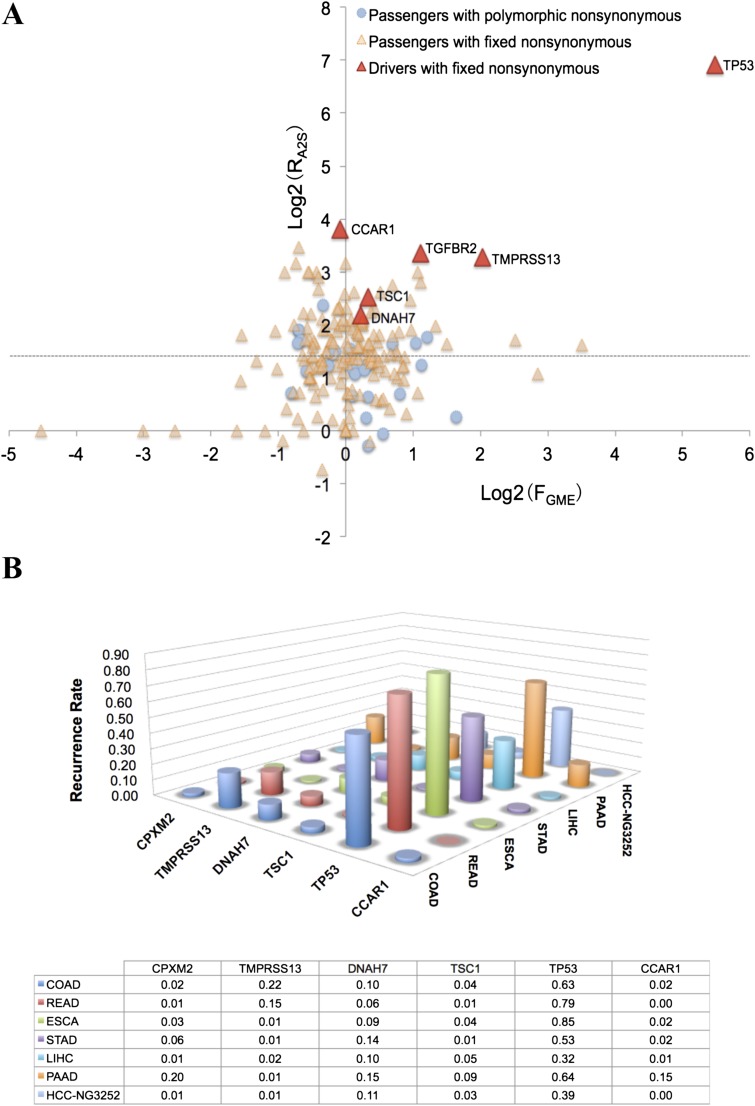
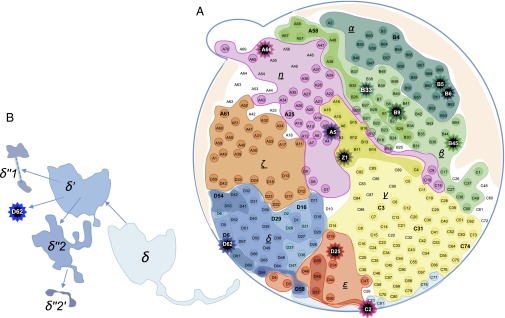
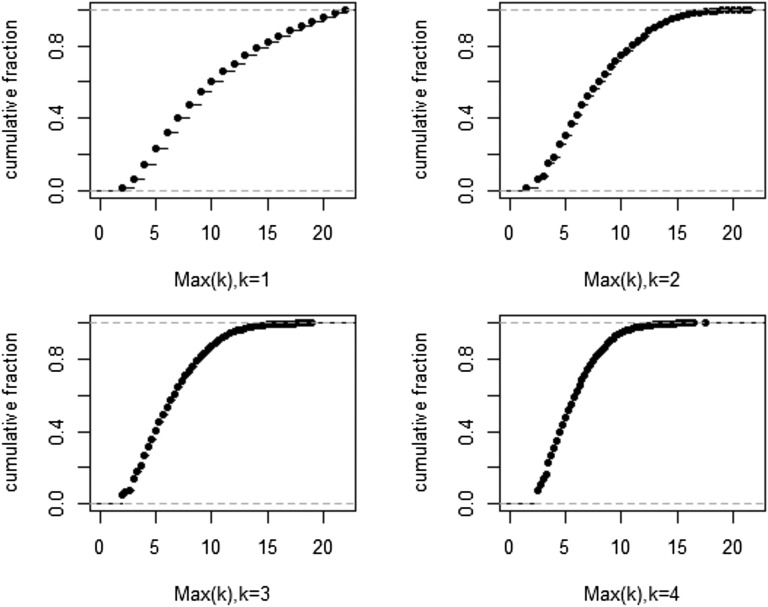
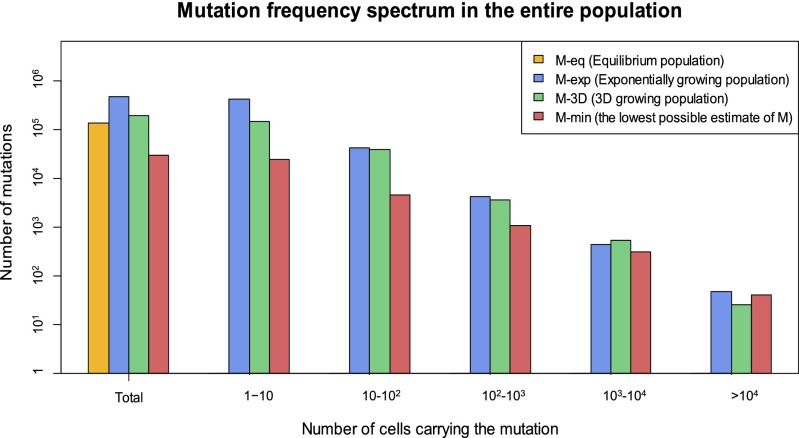
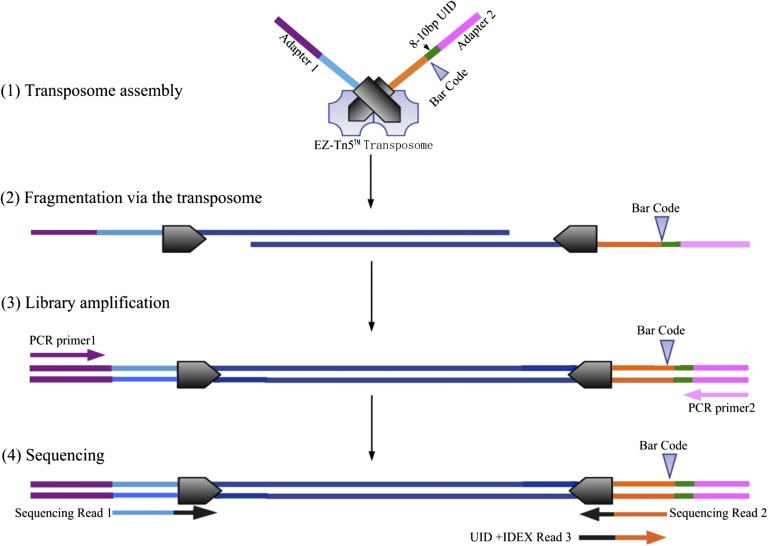
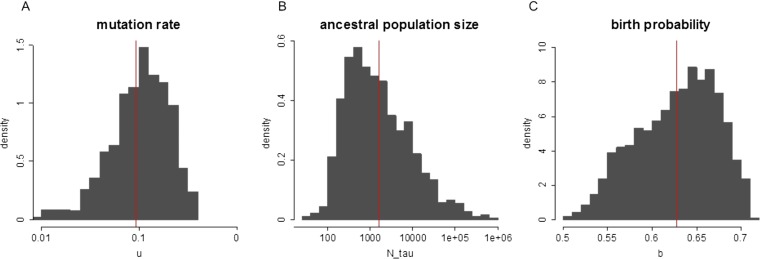
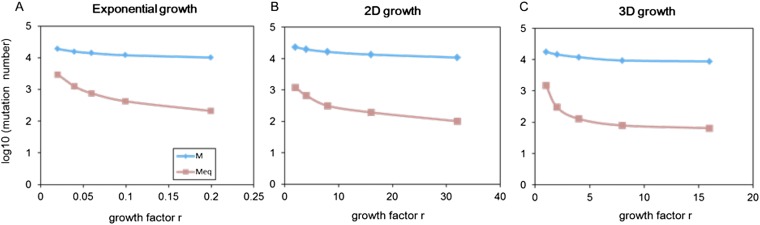
The prevailing view that the evolution of cells in a tumor is driven by Darwinian selection has never been rigorously tested. Because selection greatly affects the level of intratumor genetic diversity, it is important to assess whether intratumor evolution follows the Darwinian or the non-Darwinian mode of evolution. To provide the statistical power, many regions in a single tumor need to be sampled and analyzed much more extensively than has been attempted in previous intratumor studies. Here, from a hepatocellular carcinoma (HCC) tumor, we evaluated multiregional samples from the tumor, using either whole-exome sequencing (WES) (n = 23 samples) or genotyping (n = 286) under both the infinite-site and infinite-allele models of population genetics. In addition to the many single-nucleotide variations (SNVs) present in all samples, there were 35 "polymorphic" SNVs among samples. High genetic diversity was evident as the 23 WES samples defined 20 unique cell clones. With all 286 samples genotyped, clonal diversity agreed well with the non-Darwinian model with no evidence of positive Darwinian selection. Under the non-Darwinian model, MALL (the number of coding region mutations in the entire tumor) was estimated to be greater than 100 million in this tumor. DNA sequences reveal local diversities in small patches of cells and validate the estimation. In contrast, the genetic diversity under a Darwinian model would generally be orders of magnitude smaller. Because the level of genetic diversity will have implications on therapeutic resistance, non-Darwinian evolution should be heeded in cancer treatments even for microscopic tumors.
Keywords: cancer evolution; genetic diversity; intratumor heterogeneity; natural selection; neutral evolution.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Wen-Hsiung L. 1997. Molecular evolution (Sinauer Associates Inc., Sunderland, MA)
-
- Ewens WJ. Mathematical Population Genetics 1: Theoretical Introduction. Springer; New York: 2010.
-
- Hartl DL, Clark AG. Principle of Population Genetics. 4th Ed Sinauer; Sunderland, MA: 2006.
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. - PubMed
-
- Maley CC, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38(4):468–473. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
