

Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplification
- PMID: 26563586
- PMCID: PMC4643231
- DOI: 10.1038/srep16498
Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplification
Erratum in
-
Corrigendum: Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplification.Sci Rep. 2016 Jun 2;6:27163. doi: 10.1038/srep27163. Sci Rep. 2016. PMID: 27254246 Free PMC article. No abstract available.
Abstract
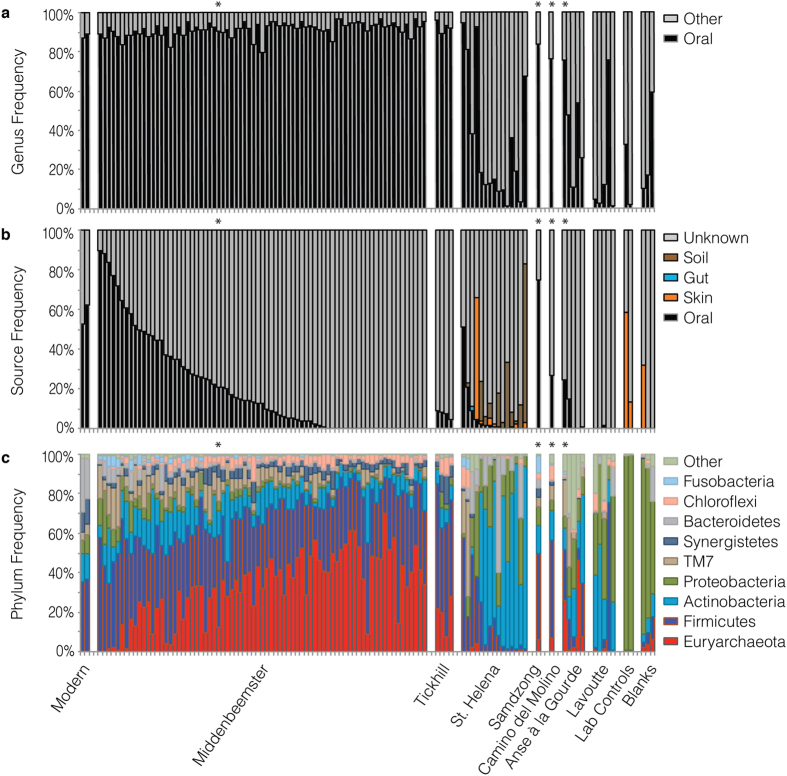
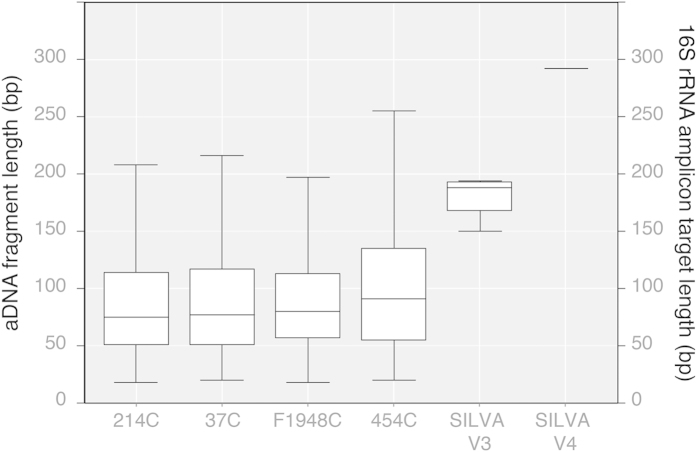
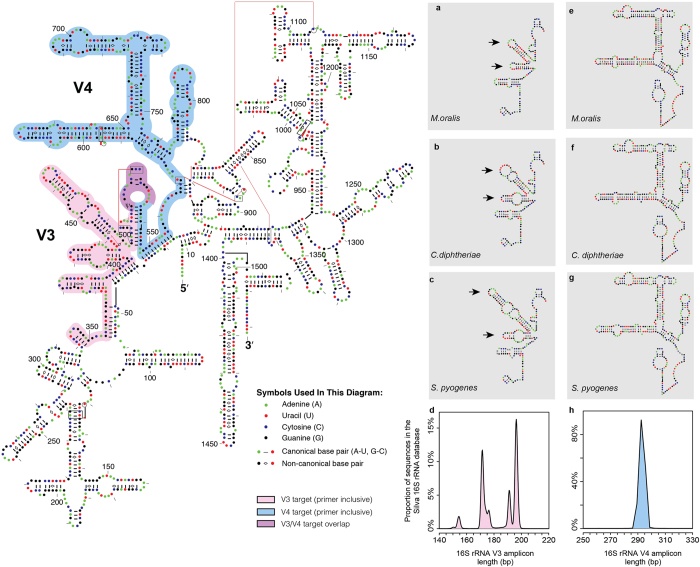
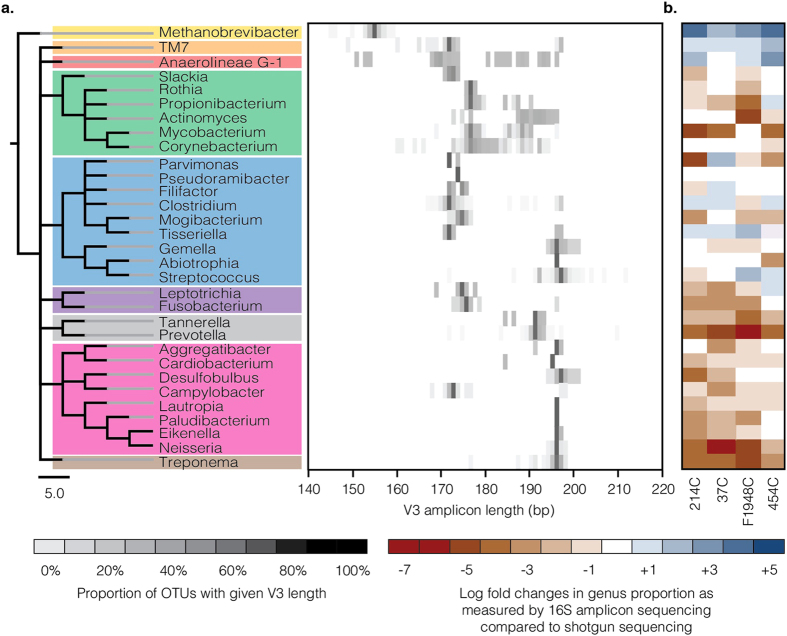
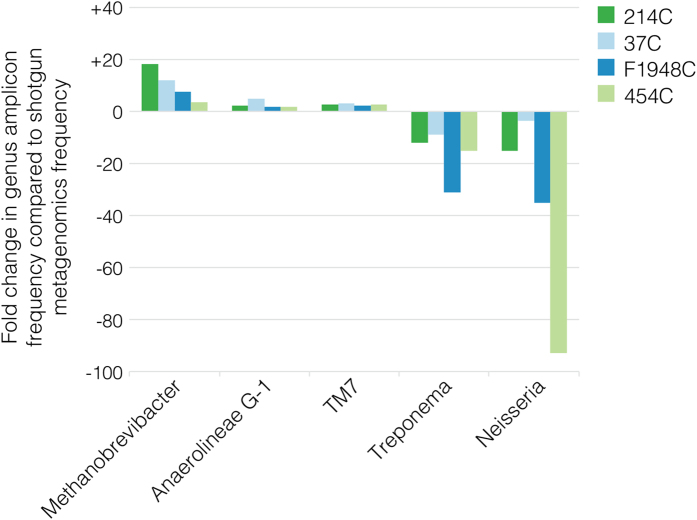
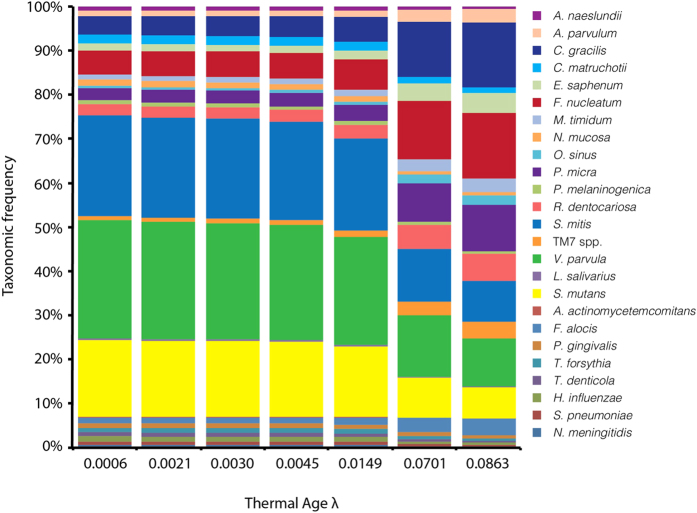
To date, characterization of ancient oral (dental calculus) and gut (coprolite) microbiota has been primarily accomplished through a metataxonomic approach involving targeted amplification of one or more variable regions in the 16S rRNA gene. Specifically, the V3 region (E. coli 341-534) of this gene has been suggested as an excellent candidate for ancient DNA amplification and microbial community reconstruction. However, in practice this metataxonomic approach often produces highly skewed taxonomic frequency data. In this study, we use non-targeted (shotgun metagenomics) sequencing methods to better understand skewed microbial profiles observed in four ancient dental calculus specimens previously analyzed by amplicon sequencing. Through comparisons of microbial taxonomic counts from paired amplicon (V3 U341F/534R) and shotgun sequencing datasets, we demonstrate that extensive length polymorphisms in the V3 region are a consistent and major cause of differential amplification leading to taxonomic bias in ancient microbiome reconstructions based on amplicon sequencing. We conclude that systematic amplification bias confounds attempts to accurately reconstruct microbiome taxonomic profiles from 16S rRNA V3 amplicon data generated using universal primers. Because in silico analysis indicates that alternative 16S rRNA hypervariable regions will present similar challenges, we advocate for the use of a shotgun metagenomics approach in ancient microbiome reconstructions.
Figures
References
-
- Lederberg J. & McCray A. ‘Ome Sweet’ Omics - A Genealogical Treasury of Words. New Sci 17 (2001).
-
- LeBlanc J. G. et al. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotech 24, 160–168 (2013). - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
