

Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit
- PMID: 26565914
- PMCID: PMC4644508
- DOI: 10.1016/j.celrep.2015.09.049
Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit
Abstract
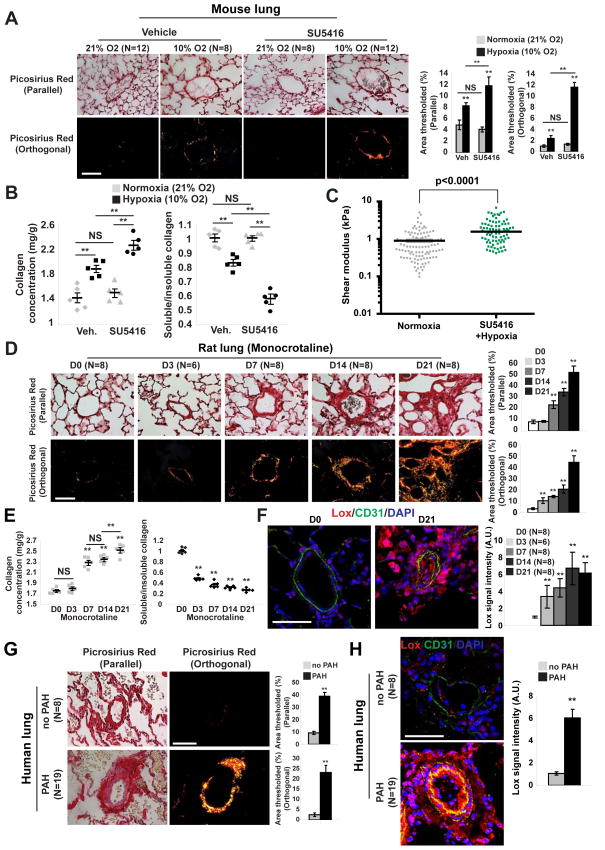
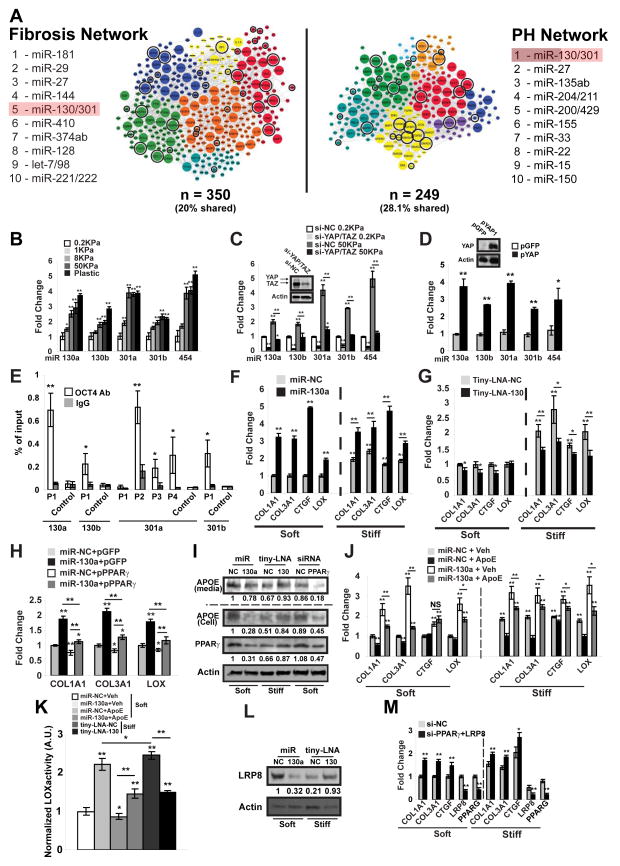
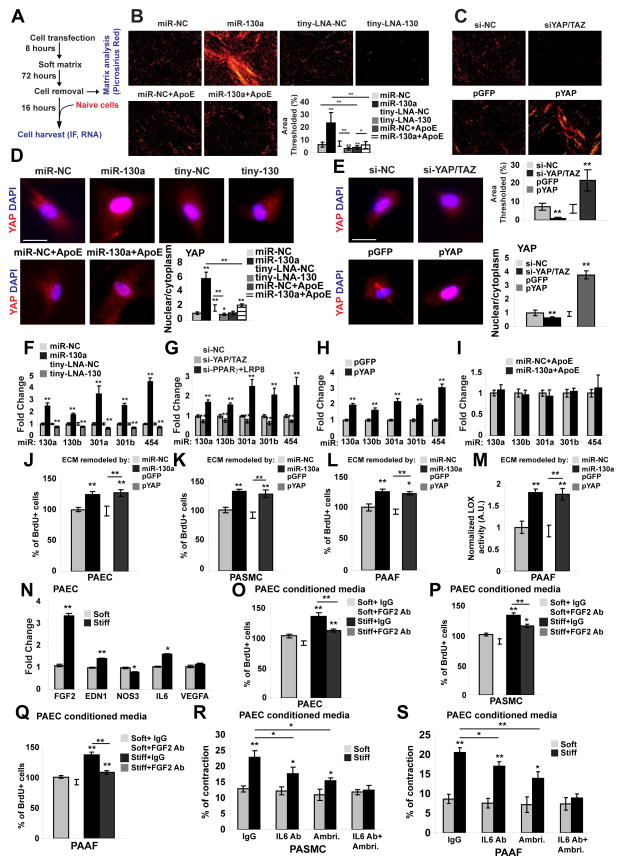
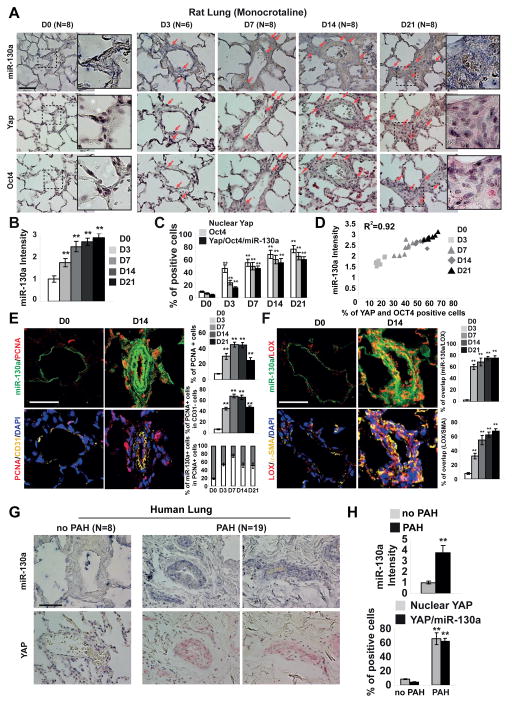
Pulmonary hypertension (PH) is a deadly vascular disease with enigmatic molecular origins. We found that vascular extracellular matrix (ECM) remodeling and stiffening are early and pervasive processes that promote PH. In multiple pulmonary vascular cell types, such ECM stiffening induced the microRNA-130/301 family via activation of the co-transcription factors YAP and TAZ. MicroRNA-130/301 controlled a PPAR?-APOE-LRP8 axis, promoting collagen deposition and LOX-dependent remodeling and further upregulating YAP/TAZ via a mechanoactive feedback loop. In turn, ECM remodeling controlled pulmonary vascular cell crosstalk via such mechanotransduction, modulation of secreted vasoactive effectors, and regulation of associated microRNA pathways. In vivo, pharmacologic inhibition of microRNA-130/301, APOE, or LOX activity ameliorated ECM remodeling and PH. Thus, ECM remodeling, as controlled by the YAP/TAZ-miR-130/301 feedback circuit, is an early PH trigger and offers combinatorial therapeutic targets for this devastating disease.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
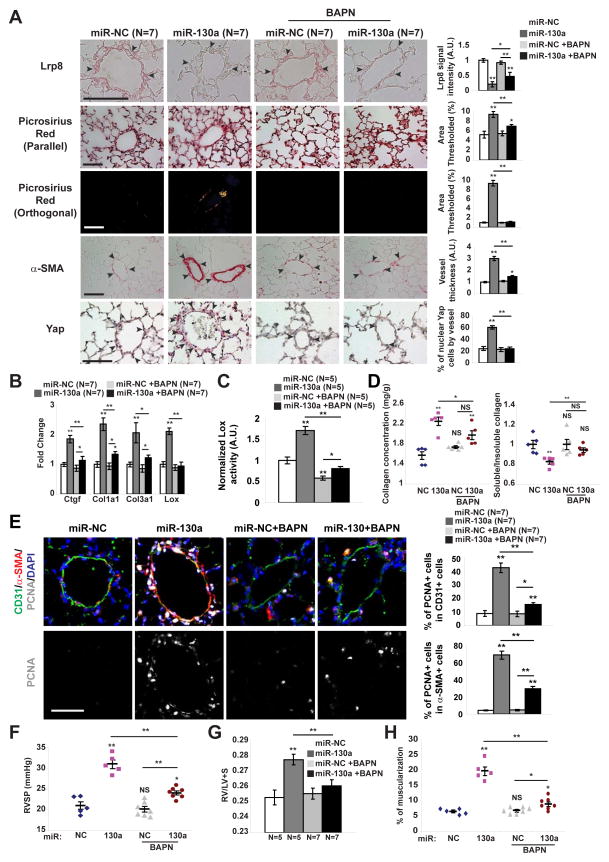
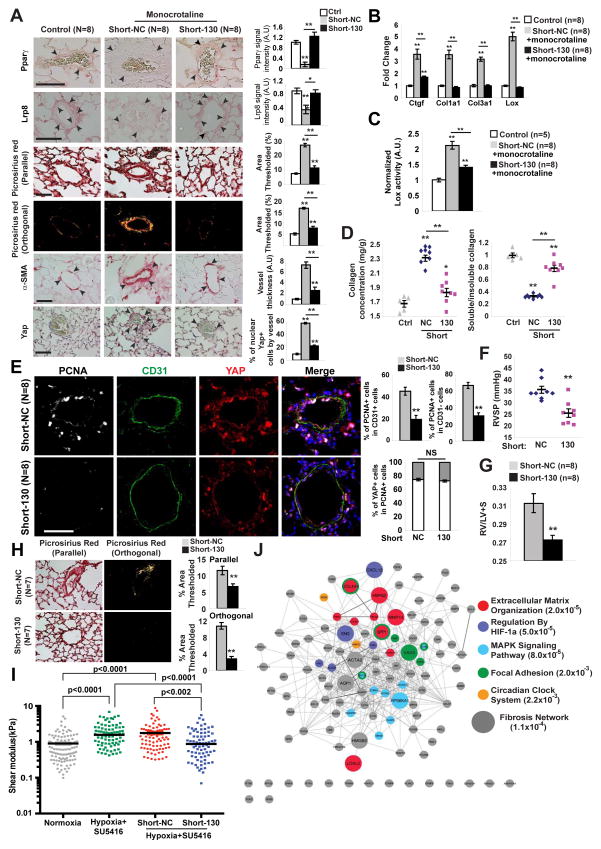
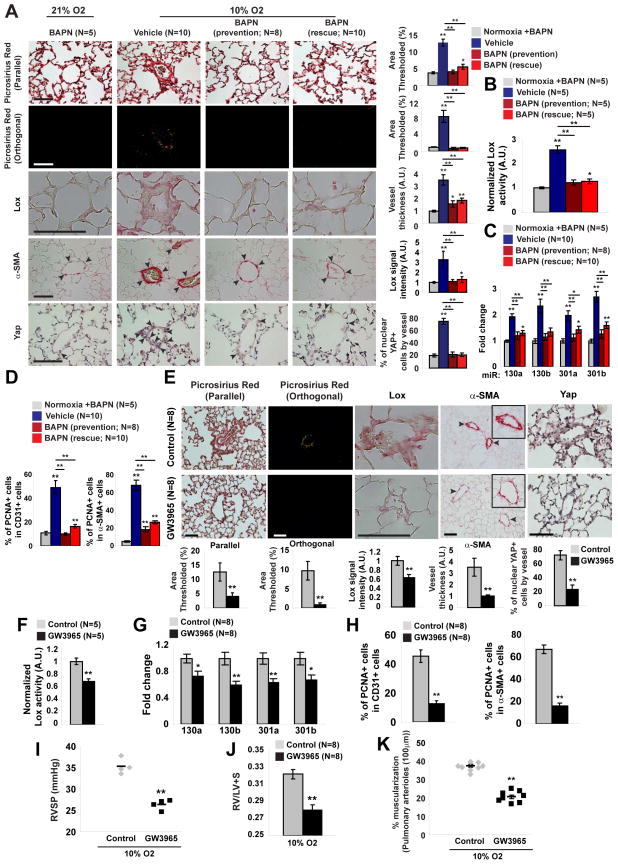
References
-
- Bi R, Bao C, Jiang L, Liu H, Yang Y, Mei J, Ding F. MicroRNA-27b plays a role in pulmonary arterial hypertension by modulating peroxisome proliferator-activated receptor gamma dependent Hsp90-eNOS signaling and nitric oxide production. Biochem Biophys Res Commun. 2015;460:469–475. - PubMed
-
- Boutet K, Montani D, Jais X, Yaici A, Sitbon O, Simonneau G, Humbert M. Therapeutic advances in pulmonary arterial hypertension. Ther Adv Respir Dis. 2008;2:249–265. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL114839/HL/NHLBI NIH HHS/United States
- R01 HL061284/HL/NHLBI NIH HHS/United States
- R01 HL122596/HL/NHLBI NIH HHS/United States
- HL67841/HL/NHLBI NIH HHS/United States
- R01 HL067841/HL/NHLBI NIH HHS/United States
- HL124021/HL/NHLBI NIH HHS/United States
- HL096834/HL/NHLBI NIH HHS/United States
- K08 HL096834/HL/NHLBI NIH HHS/United States
- R03 HL115106/HL/NHLBI NIH HHS/United States
- R01 HL124021/HL/NHLBI NIH HHS/United States
- R01 HL060190/HL/NHLBI NIH HHS/United States
- T32 HD049303/HD/NICHD NIH HHS/United States
- K08 HL105536/HL/NHLBI NIH HHS/United States
- HL61284/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
