

New Insights into the Pathogenesis of IgA Nephropathy
- PMID: 26568951
- PMCID: PMC4640461
- DOI: 10.1159/000382134
New Insights into the Pathogenesis of IgA Nephropathy
Abstract
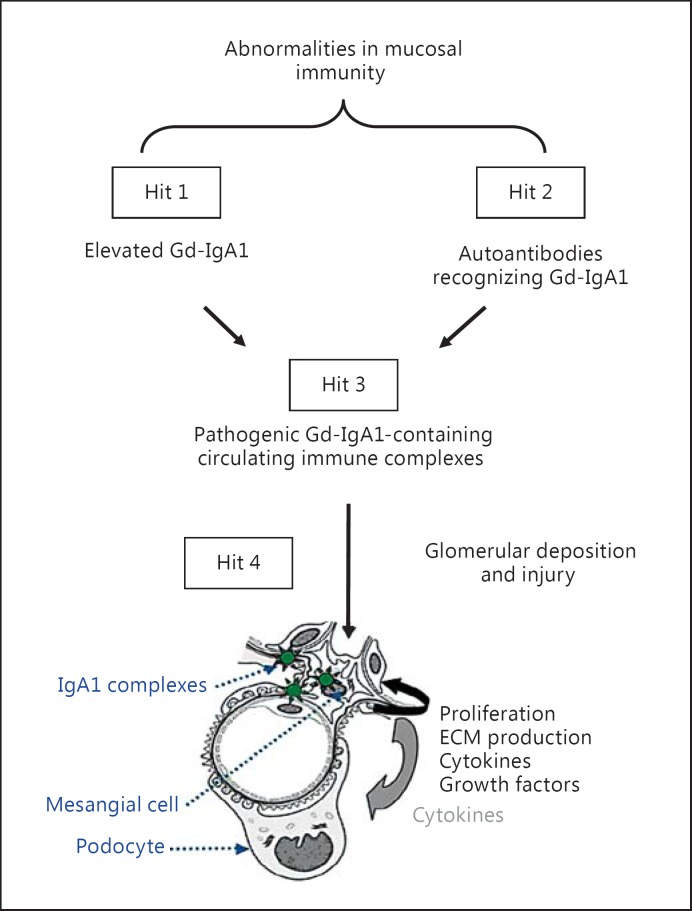
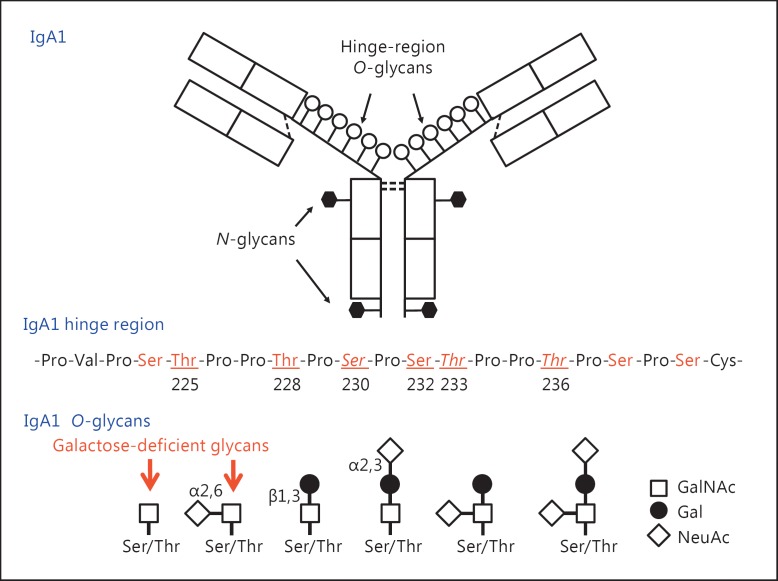
Background: IgA nephropathy, a frequent cause of end-stage renal disease, is an autoimmune disease wherein immune complexes consisting of IgA1 with galactose-deficient O-glycans (autoantigen) and anti-glycan autoantibodies deposit in glomeruli and induce renal injury. Multiple genetic loci associated with disease risk have been identified. The prevalence of risk alleles varies geographically, highest in eastern Asia and northern Europe, fewer in other parts of Europe and North America, and the least in Africa. IgA nephropathy is diagnosed from pathological assessment of a renal biopsy specimen. Currently, therapy is not disease-targeted but rather is focused on maintaining control of blood pressure and proteinuria, ideally with suppression of angiotensin II. Possible additional approaches differ between countries. Disease-specific therapy as well as new tools for diagnosis, prognosis, and assessment of responses to therapy are needed.
Summary: Glycosylation pathways associated with aberrant O-glycosylation of IgA1 and, thus, production of autoantigen, have been identified. Furthermore, unique characteristics of the autoantibodies in IgA nephropathy have been uncovered. Many of these biochemical features are shared by patients with IgA nephropathy and Henoch-Schönlein purpura nephritis, suggesting that the two diseases may represent opposite ends of a spectrum of a disease process. Understanding the molecular mechanisms involved in formation of pathogenic IgA1-containing immune complexes will enable development of disease-specific therapies as well as diagnostic and prognostic biomarkers.
Key messages: IgA nephropathy is an autoimmune disease caused by glomerular deposition of nephritogenic circulating immune complexes consisting of galactose-deficient IgA1 (autoantigen) bound by anti-glycan autoantibodies. A better understanding of the multi-step process of pathogenesis of IgA nephropathy and the genetic and environmental contributing factors will lead to development of biomarkers to identify patients with progressive disease who would benefit from a future disease-specific therapy.
Keywords: Autoantibodies; IgA nephropathy; O-glycans; galactose deficiency; immune complexes.
Figures
References
-
- D'Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Quart J Med. 1987;64:709–727. - PubMed
-
- Julian BA, Waldo FB, Rifai A, Mestecky J. IgA nephropathy, the most common glomerulonephritis worldwide. A neglected disease in the United States? Am J Med. 1988;84:129–132. - PubMed
-
- Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368:2402–2414. - PubMed
-
- Berger J, Hinglais N. Les dépôts intercapillaires d'IgA-IgG (Intercapillary deposits of IgA-IgG) J Urol Nephrol. 1968;74:694–695. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
