

Assessment of the Utility of Whole Genome Sequencing of Measles Virus in the Characterisation of Outbreaks
- PMID: 26569100
- PMCID: PMC4646484
- DOI: 10.1371/journal.pone.0143081
Assessment of the Utility of Whole Genome Sequencing of Measles Virus in the Characterisation of Outbreaks
Abstract
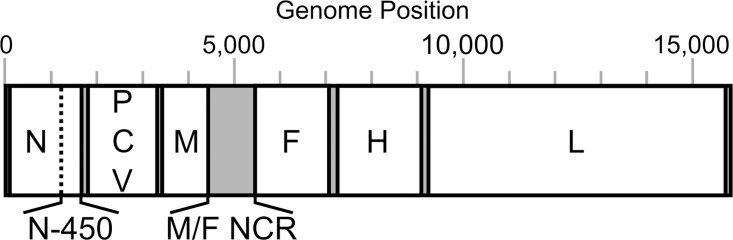
Background: Measles is a highly infectious disease caused by measles virus (MeV). Despite the availability of a safe and cost-effective vaccine, measles is one of the world-leading causes of death in young children. Within Europe, there is a target for eliminating endemic measles in 2015, with molecular epidemiology required on 80% of cases for inclusion/exclusion of outbreak transmission chains. Currently, MeV is genotyped on the basis of a 450 nucleotide region of the nucleoprotein gene (N-450) and the hemagglutinin gene (H). However, this is not sufficiently informative for distinguishing endemic from imported MeV. We have developed an amplicon-based method for obtaining whole genome sequences (WGS) using NGS or Sanger methodologies from cell culture isolates or oral fluid specimens, and have sequenced over 60 samples, including 42 from the 2012 outbreak in the UK.
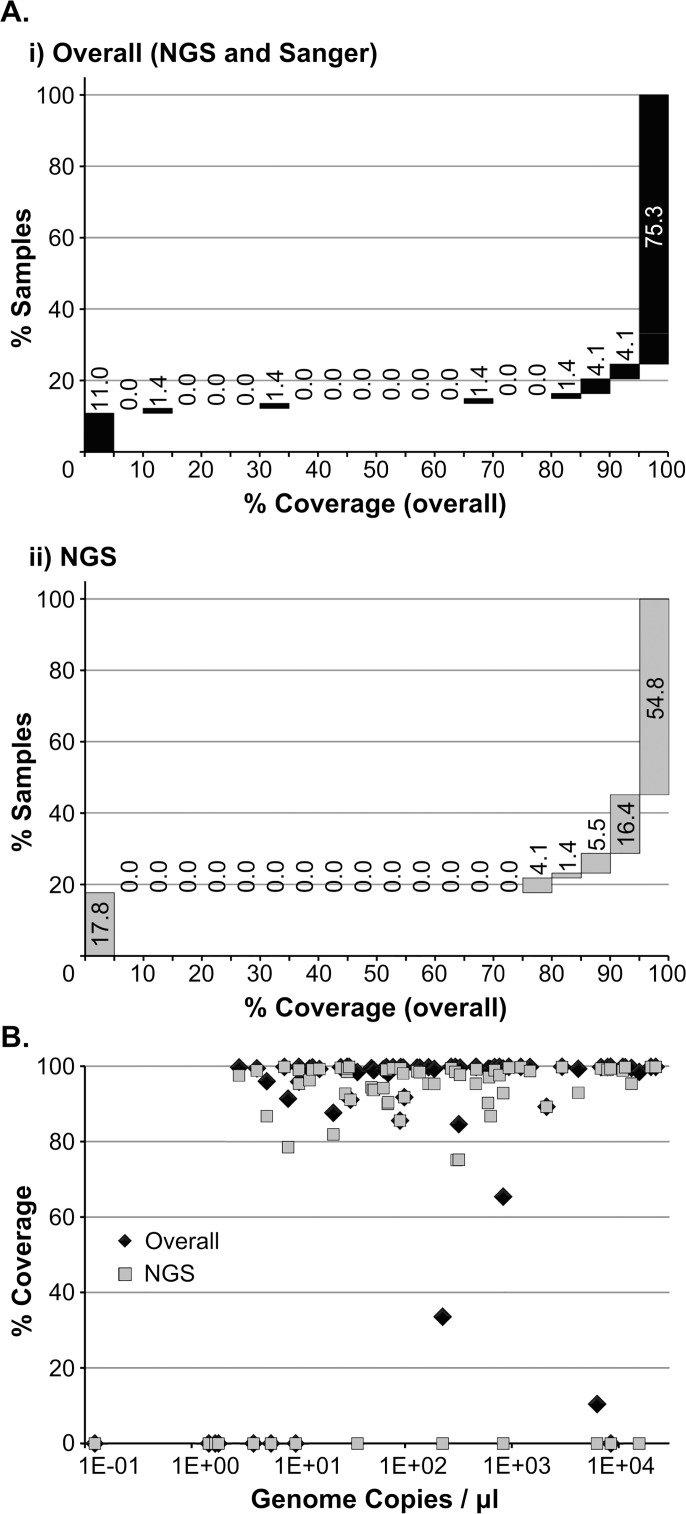
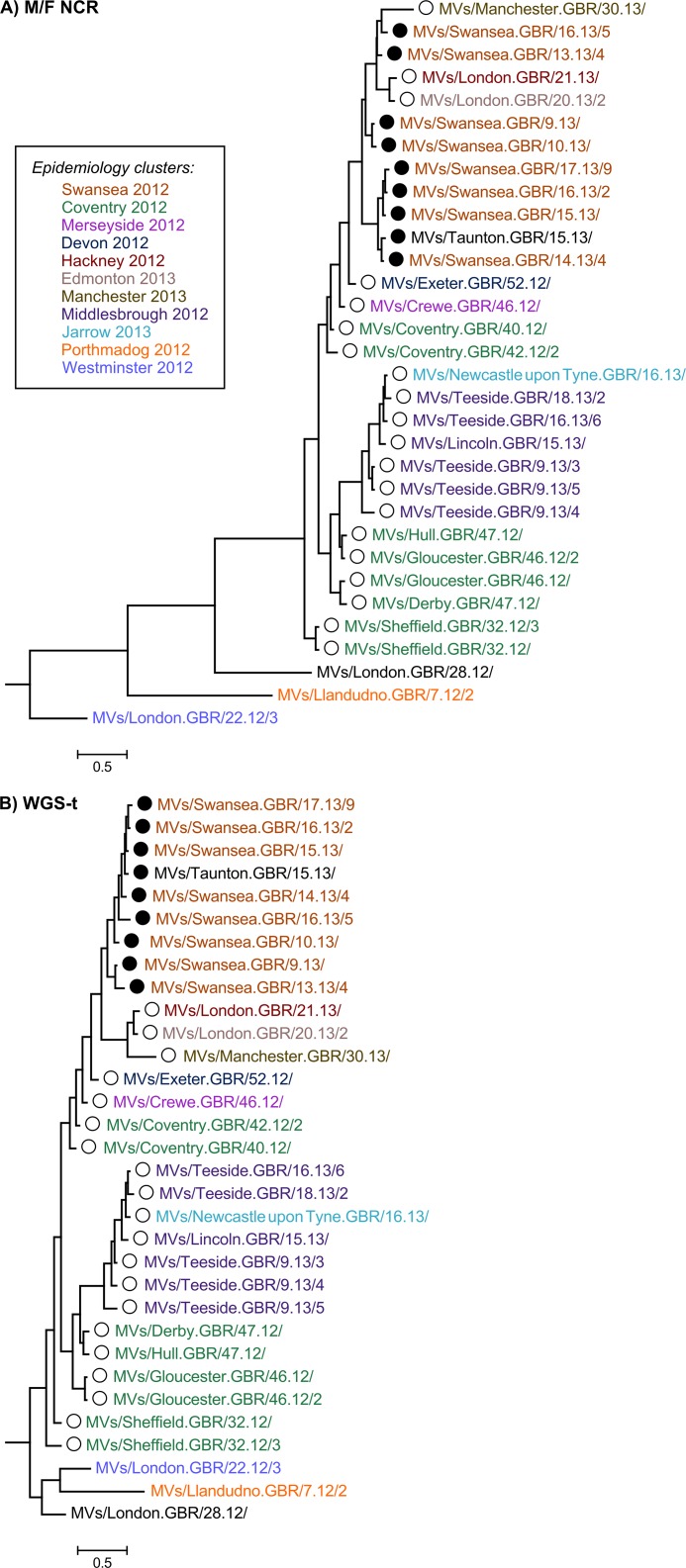
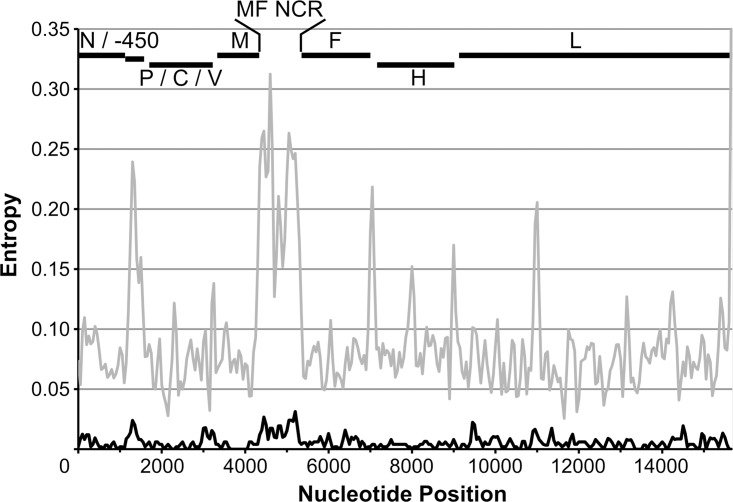
Results: Overall, NGS coverage was over 90% for approximately 71% of the samples tested. Analysis of 32 WGS excluding 3' and 5' termini (WGS-t) obtained from the outbreak indicates that the single nucleotide difference found between the two major groups of N-450 sequences detected during the outbreak is most likely a result of stochastic viral mutation during endemic transmission rather than of multiple importation events: earlier strains appear to have evolved into two distinct strain clusters in 2013, one containing strains with both outbreak-associated N-450 sequences. Additionally, phylogenetic analysis of each genomic region of MeV for the strains in this study suggests that the most information is acquired from the non-coding region located between the matrix and fusion protein genes (M/F NCR) and the N-450 genotyping sequence, an observation supported by entropy analysis across genotypes.
Conclusions: We suggest that both M/F NCR and WGS-t could be used to complement the information from classical epidemiology and N-450 sequencing to address specific questions in the context of measles elimination.
Conflict of interest statement
Figures


References
-
- van den Ent MM, Brown DW, Hoekstra EJ, Christie A, Cochi SL. Measles mortality reduction contributes substantially to reduction of all cause mortality among children less than five years of age, 1990–2008. JInfectDis. 2011;204 Suppl 1:S18–S23. jir081 [pii]; - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
