

Parametrization of Backbone Flexibility in a Coarse-Grained Force Field for Proteins (COFFDROP) Derived from All-Atom Explicit-Solvent Molecular Dynamics Simulations of All Possible Two-Residue Peptides
- PMID: 26574429
- PMCID: PMC4658516
- DOI: 10.1021/acs.jctc.5b00038
Parametrization of Backbone Flexibility in a Coarse-Grained Force Field for Proteins (COFFDROP) Derived from All-Atom Explicit-Solvent Molecular Dynamics Simulations of All Possible Two-Residue Peptides
Abstract
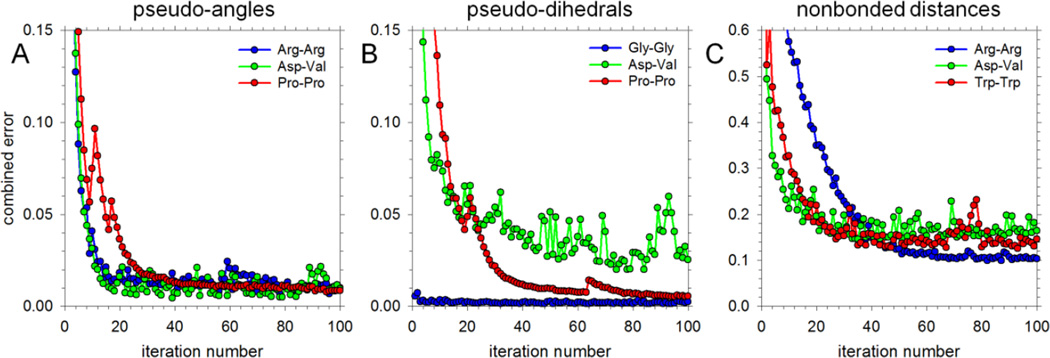
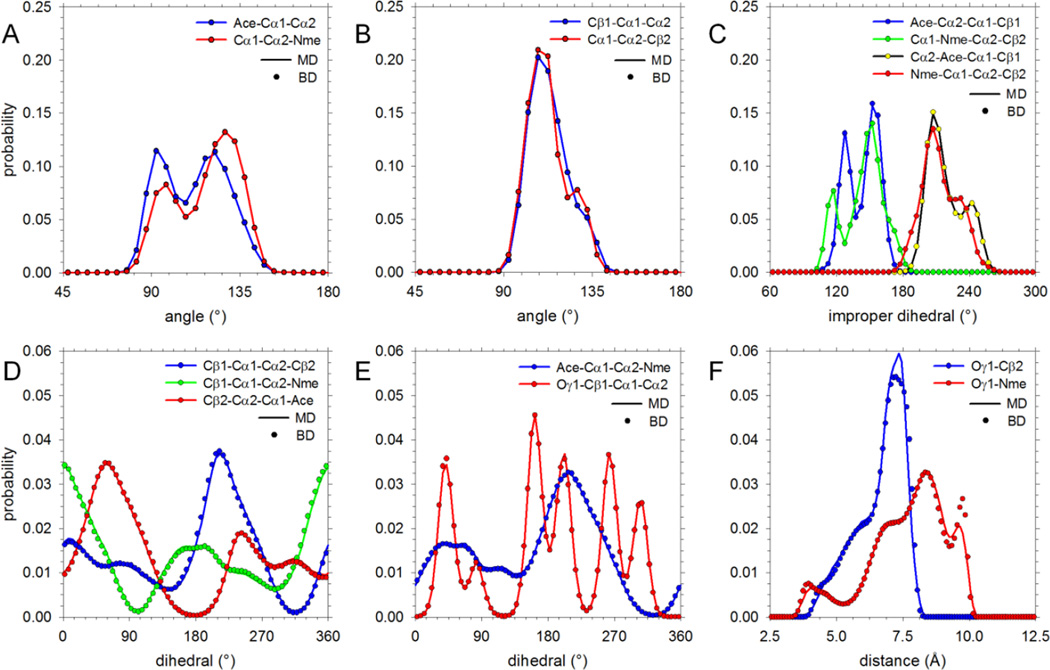
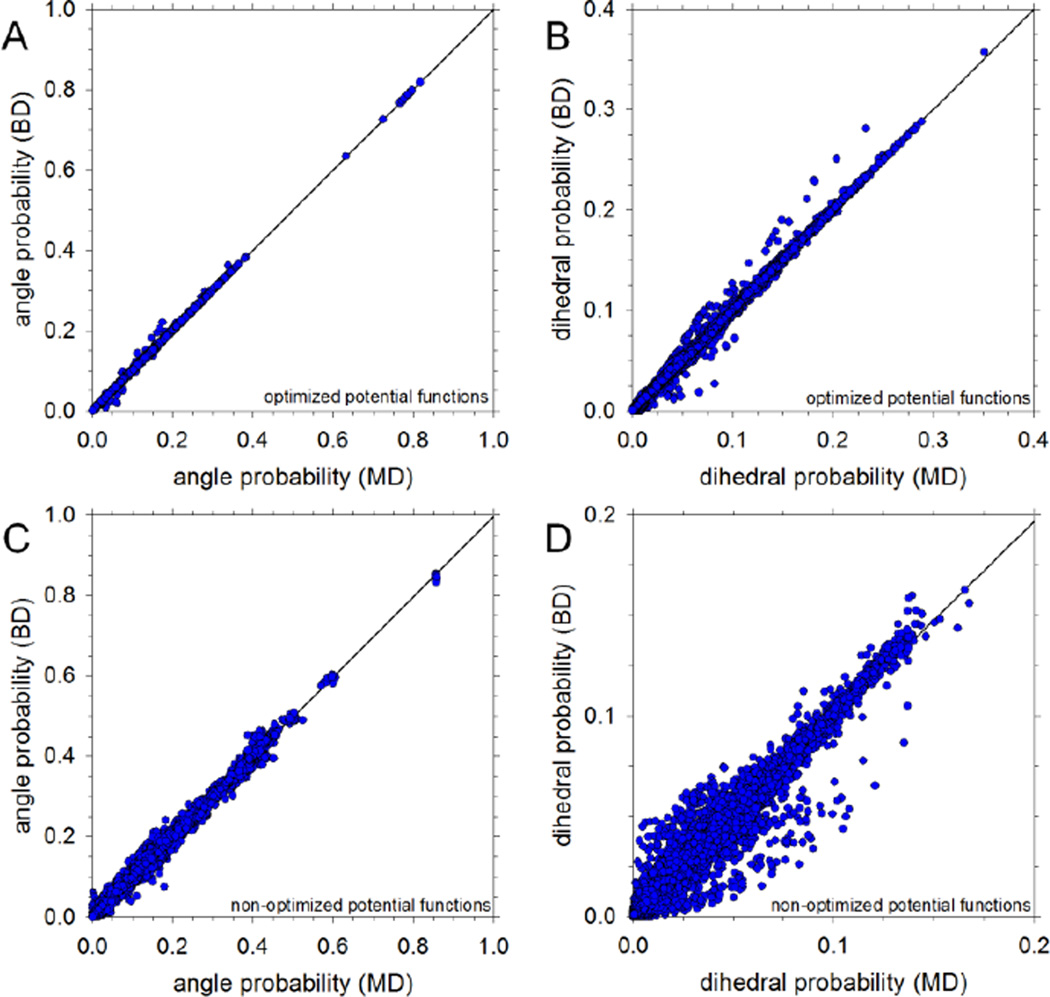
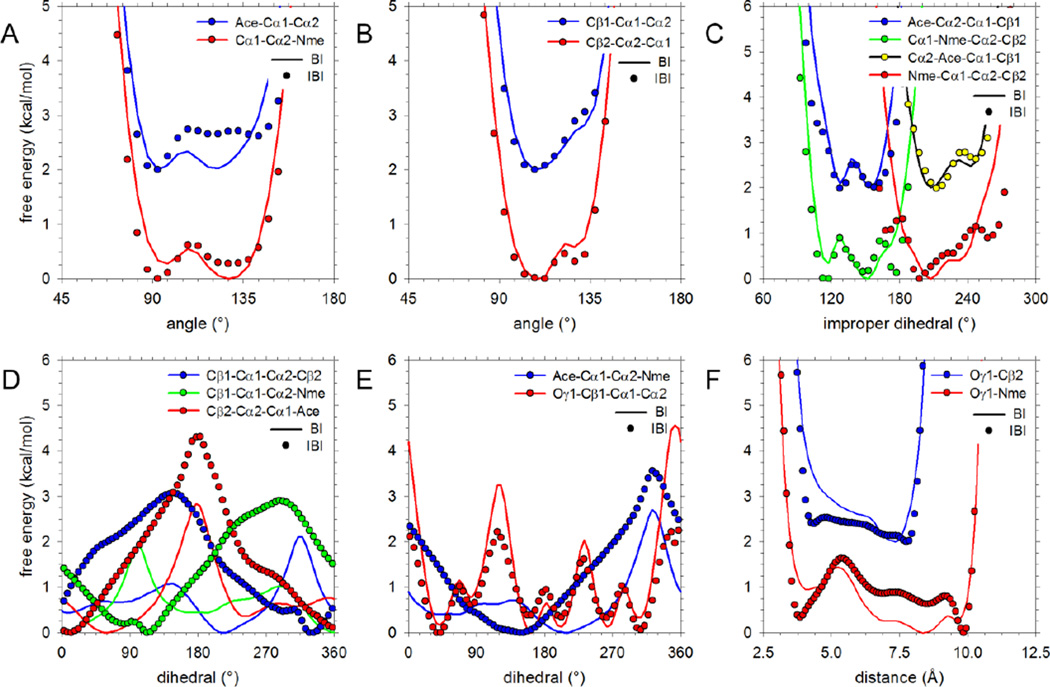
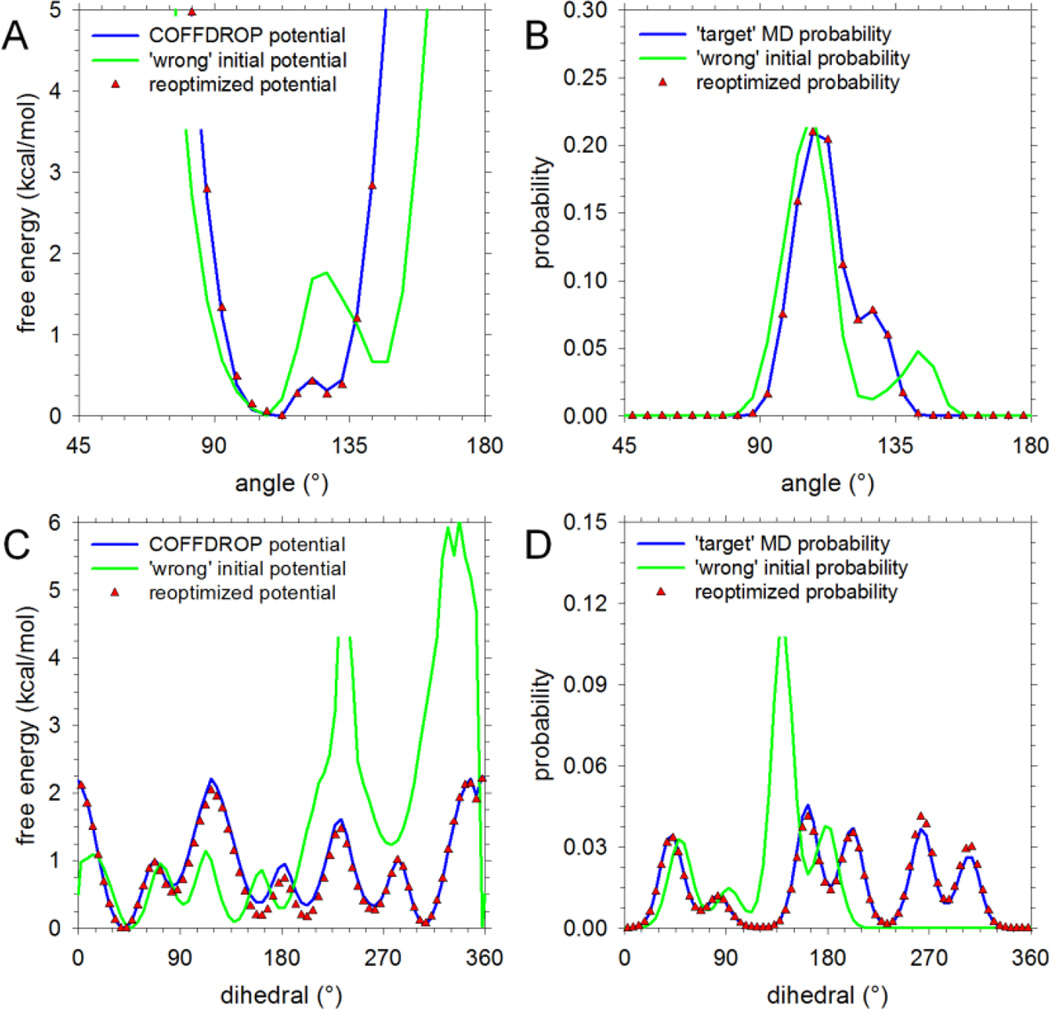
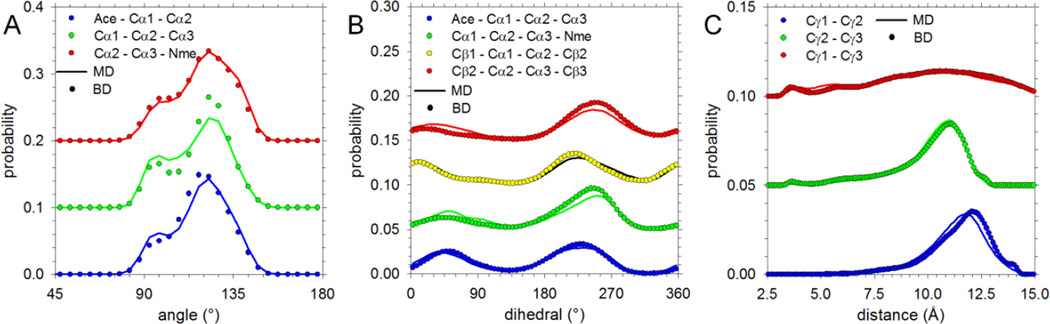
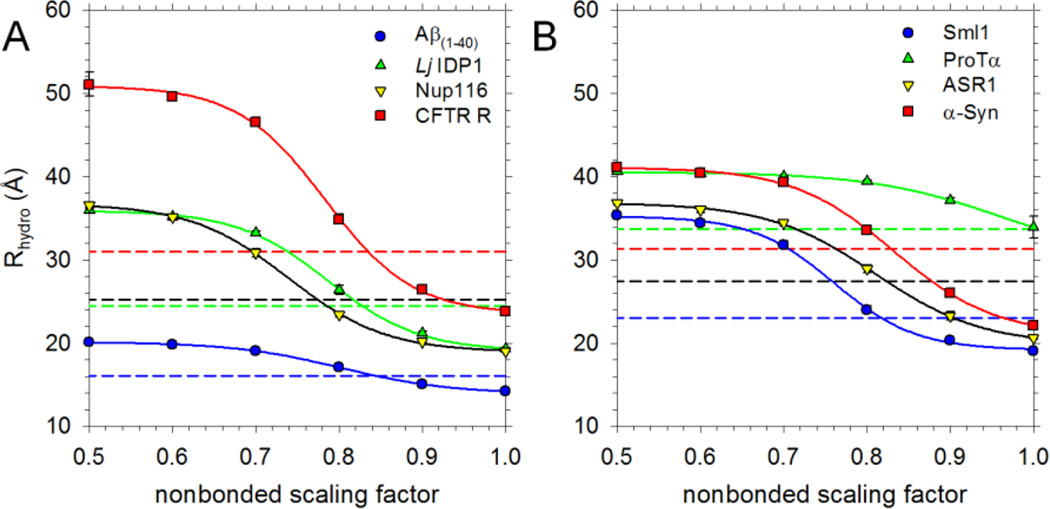
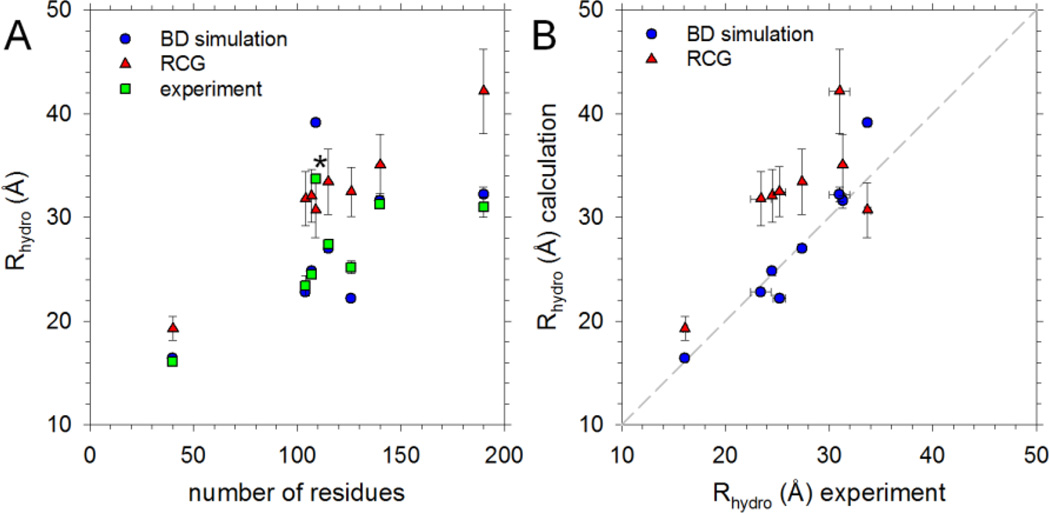
Recently, we reported the parametrization of a set of coarse-grained (CG) nonbonded potential functions, derived from all-atom explicit-solvent molecular dynamics (MD) simulations of amino acid pairs and designed for use in (implicit-solvent) Brownian dynamics (BD) simulations of proteins; this force field was named COFFDROP (COarse-grained Force Field for Dynamic Representations Of Proteins). Here, we describe the extension of COFFDROP to include bonded backbone terms derived from fitting to results of explicit-solvent MD simulations of all possible two-residue peptides containing the 20 standard amino acids, with histidine modeled in both its protonated and neutral forms. The iterative Boltzmann inversion (IBI) method was used to optimize new CG potential functions for backbone-related terms by attempting to reproduce angle, dihedral, and distance probability distributions generated by the MD simulations. In a simple test of the transferability of the extended force field, the angle, dihedral, and distance probability distributions obtained from BD simulations of 56 three-residue peptides were compared to results from corresponding explicit-solvent MD simulations. In a more challenging test of the COFFDROP force field, it was used to simulate eight intrinsically disordered proteins and was shown to quite accurately reproduce the experimental hydrodynamic radii (Rhydro), provided that the favorable nonbonded interactions of the force field were uniformly scaled downward in magnitude. Overall, the results indicate that the COFFDROP force field is likely to find use in modeling the conformational behavior of intrinsically disordered proteins and multidomain proteins connected by flexible linkers.
Figures
Similar articles
-
COFFDROP: A Coarse-Grained Nonbonded Force Field for Proteins Derived from All-Atom Explicit-Solvent Molecular Dynamics Simulations of Amino Acids.J Chem Theory Comput. 2014 Nov 11;10(11):5178-5194. doi: 10.1021/ct5006328. Epub 2014 Oct 7. J Chem Theory Comput. 2014. PMID: 25400526 Free PMC article.
-
Hybrid simulations: combining atomistic and coarse-grained force fields using virtual sites.Phys Chem Chem Phys. 2011 Jun 14;13(22):10437-48. doi: 10.1039/c0cp02981e. Epub 2011 Apr 15. Phys Chem Chem Phys. 2011. PMID: 21494747
-
Self-assembling dipeptides: conformational sampling in solvent-free coarse-grained simulation.Phys Chem Chem Phys. 2009 Mar 28;11(12):2077-86. doi: 10.1039/b818144f. Epub 2009 Feb 2. Phys Chem Chem Phys. 2009. PMID: 19280018
-
Developments and Applications of Coil-Library-Based Residue-Specific Force Fields for Molecular Dynamics Simulations of Peptides and Proteins.J Chem Theory Comput. 2019 May 14;15(5):2761-2773. doi: 10.1021/acs.jctc.8b00794. Epub 2019 Apr 8. J Chem Theory Comput. 2019. PMID: 30620582 Review.
-
Accurate coarse grained models for protein association and recognition.Adv Protein Chem Struct Biol. 2025;145:1-21. doi: 10.1016/bs.apcsb.2024.11.011. Epub 2025 Apr 23. Adv Protein Chem Struct Biol. 2025. PMID: 40324844 Review.
Cited by
-
Direct Comparison of Amino Acid and Salt Interactions with Double-Stranded and Single-Stranded DNA from Explicit-Solvent Molecular Dynamics Simulations.J Chem Theory Comput. 2017 Apr 11;13(4):1794-1811. doi: 10.1021/acs.jctc.6b00883. Epub 2017 Mar 24. J Chem Theory Comput. 2017. PMID: 28288277 Free PMC article.
-
Investigating Intrinsically Disordered Proteins With Brownian Dynamics.Front Mol Biosci. 2022 Jun 8;9:898838. doi: 10.3389/fmolb.2022.898838. eCollection 2022. Front Mol Biosci. 2022. PMID: 35755809 Free PMC article.
-
Tabulation as a high-resolution alternative to coarse-graining protein interactions: Initial application to virus capsid subunits.J Chem Phys. 2015 Dec 28;143(24):243159. doi: 10.1063/1.4938479. J Chem Phys. 2015. PMID: 26723644 Free PMC article.
-
A Stochastic Multiscale Model of Cardiac Thin Filament Activation Using Brownian-Langevin Dynamics.Biophys J. 2019 Dec 17;117(12):2255-2272. doi: 10.1016/j.bpj.2019.08.003. Epub 2019 Aug 9. Biophys J. 2019. PMID: 31547973 Free PMC article.
-
CAMELOT: A machine learning approach for coarse-grained simulations of aggregation of block-copolymeric protein sequences.J Chem Phys. 2015 Dec 28;143(24):243123. doi: 10.1063/1.4935066. J Chem Phys. 2015. PMID: 26723608 Free PMC article.
References
-
- Noid WG. Perspective: coarse-grained models for biomolecular systems. J. Chem. Phys. 2013;139:090901. - PubMed
-
- Brini E, Algaer EA, Ganguly P, Li C, Rodriguez-Ropero F, van der Vegt NF. Systematic coarse-graining methods for soft matter simulations - a review. Soft Matter. 2013;9:2108–2119.
-
- Saunders MG, Voth GA. Coarse-graining methods for computational biology. Annu Rev. Biophys. 2013;42:73–93. - PubMed
-
- Riniker S, Allison JR, van Gunsteren WF. On developing coarse-grained models for biomolecular simulation: a review. Phys. Chem. Chem. Phys. 2012;14:12423–12430. - PubMed
-
- Takada S. Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct. Biol. 2012;22:130–137. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources