

Cross-biome comparison of microbial association networks
- PMID: 26579106
- PMCID: PMC4621437
- DOI: 10.3389/fmicb.2015.01200
Cross-biome comparison of microbial association networks
Abstract
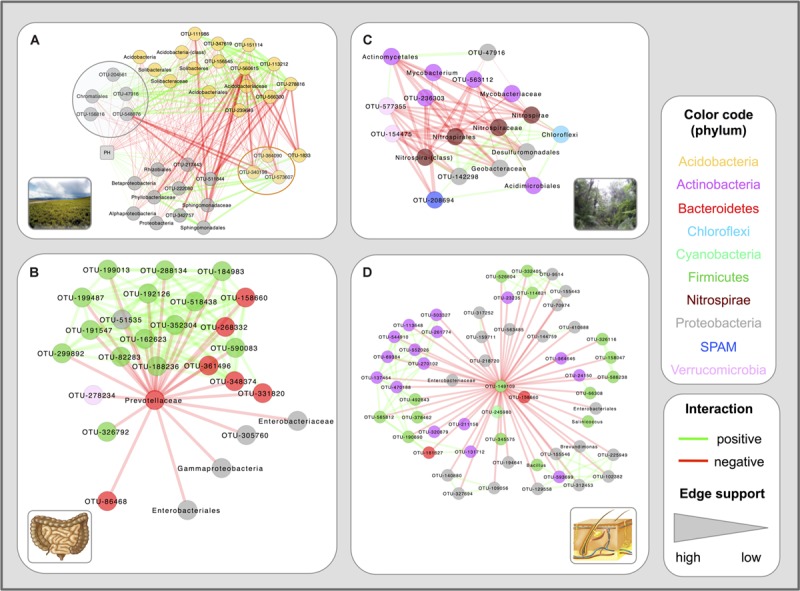
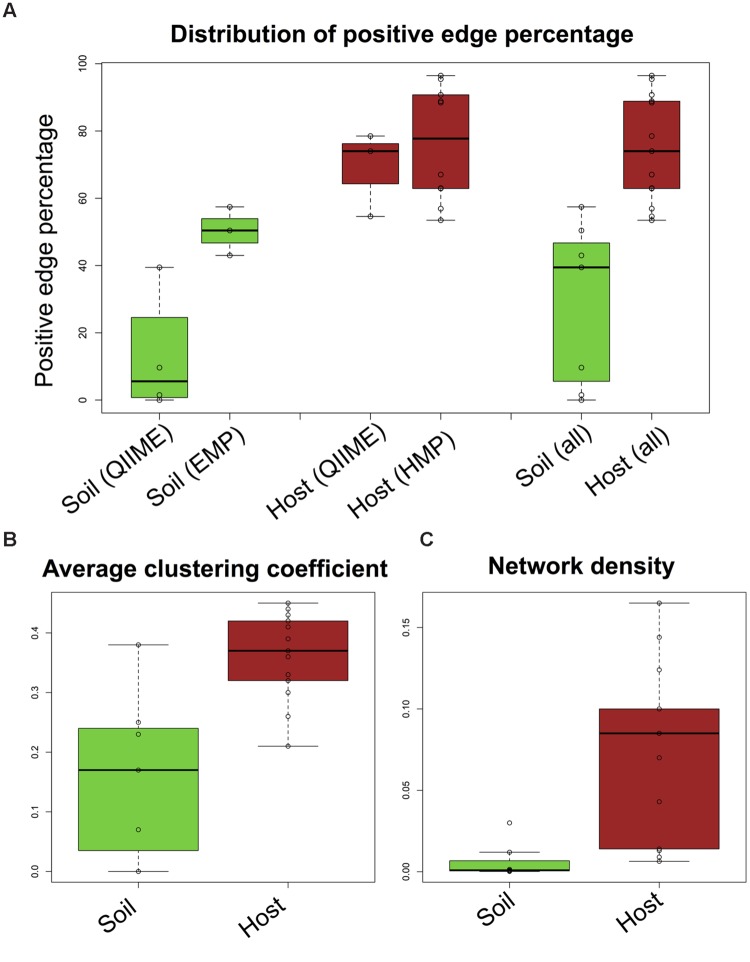
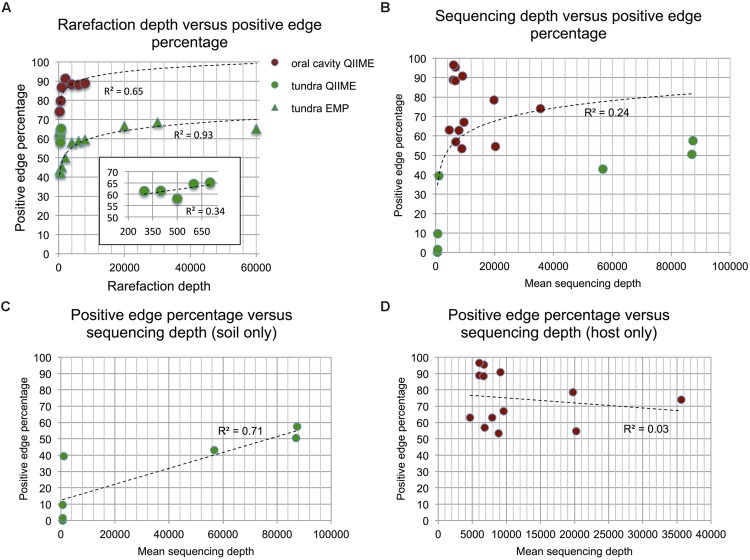
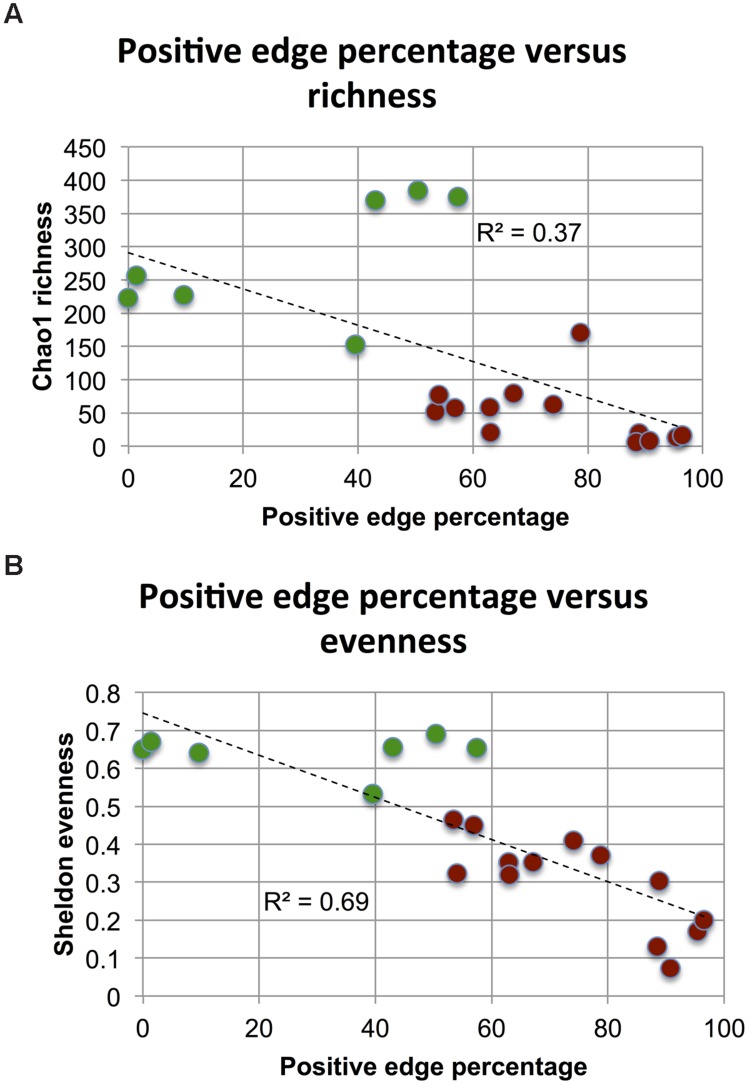
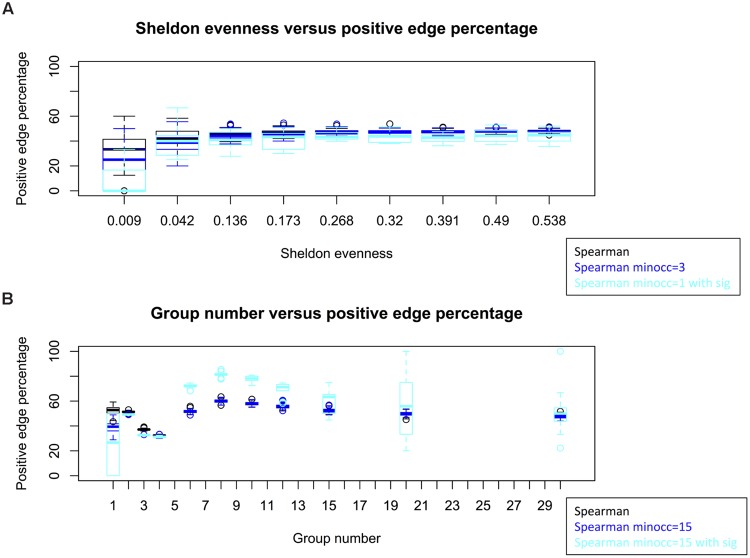
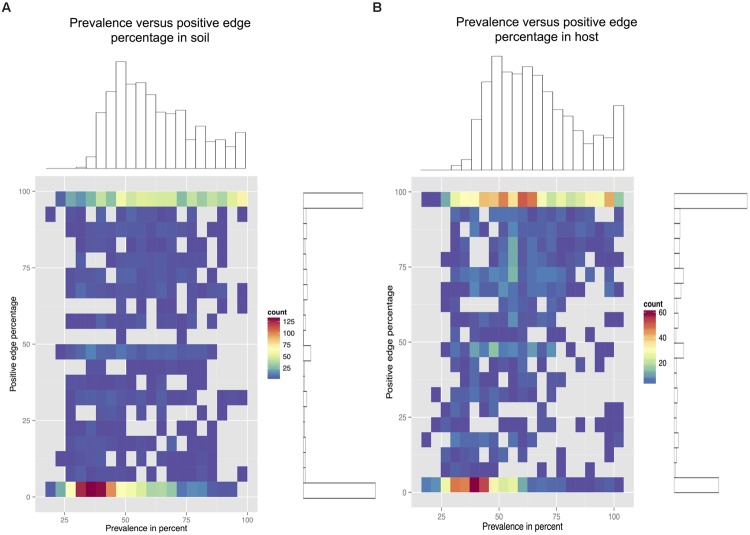
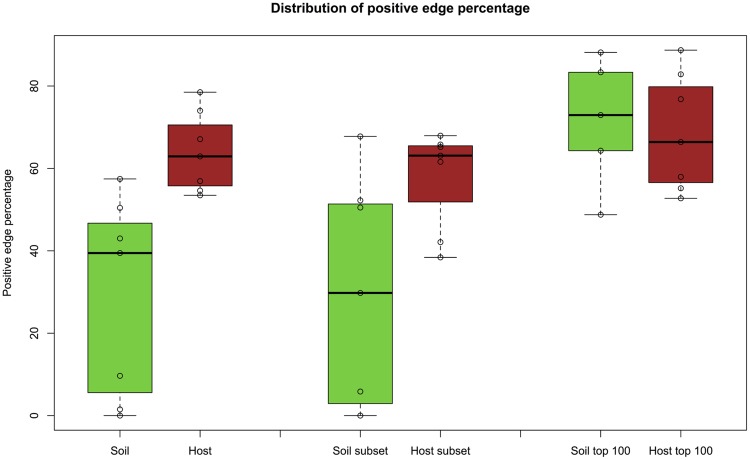
Clinical and environmental meta-omics studies are accumulating an ever-growing amount of microbial abundance data over a wide range of ecosystems. With a sufficiently large sample number, these microbial communities can be explored by constructing and analyzing co-occurrence networks, which detect taxon associations from abundance data and can give insights into community structure. Here, we investigate how co-occurrence networks differ across biomes and which other factors influence their properties. For this, we inferred microbial association networks from 20 different 16S rDNA sequencing data sets and observed that soil microbial networks harbor proportionally fewer positive associations and are less densely interconnected than host-associated networks. After excluding sample number, sequencing depth and beta-diversity as possible drivers, we found a negative correlation between community evenness and positive edge percentage. This correlation likely results from a skewed distribution of negative interactions, which take place preferentially between less prevalent taxa. Overall, our results suggest an under-appreciated role of evenness in shaping microbial association networks.
Keywords: 16S rDNA sequencing; co-occurrence; evenness; microbial communities; network comparison; positive edge percentage.
Figures
References
-
- Aderhold A., Husmeier D., Lennon J. J., Beale C. M., Smith V. A. (2012). Hierarchical Bayesian models in ecology: reconstructing species interaction networks from non-homogeneous species abundance data. Ecol. Inform. 11 55–64. 10.1016/j.ecoinf.2012.05.002 - DOI
-
- Aitchison J. (2003). “A concise guide to compositional data analysis,” in Proceedings of the 2nd Compositional Data Analysis Workshop Girona.
-
- Alatalo R. V. (1981). Problems in the measurement of evenness in ecology. Oikos 37 199–204. 10.2307/3544465 - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources