

Leveraging Distant Relatedness to Quantify Human Mutation and Gene-Conversion Rates
- PMID: 26581902
- PMCID: PMC4678427
- DOI: 10.1016/j.ajhg.2015.10.006
Leveraging Distant Relatedness to Quantify Human Mutation and Gene-Conversion Rates
Abstract
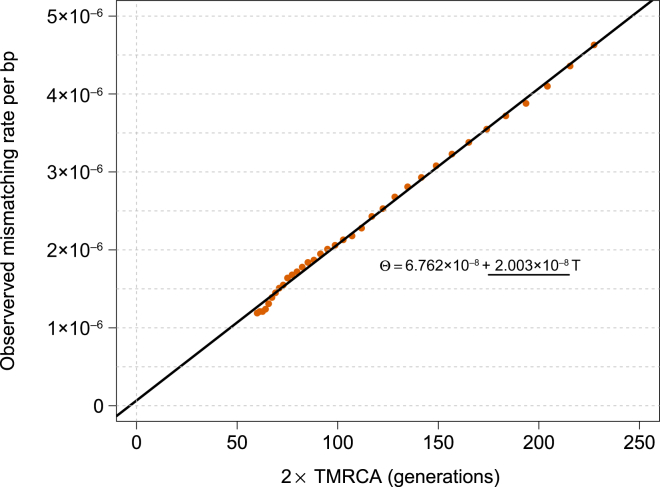
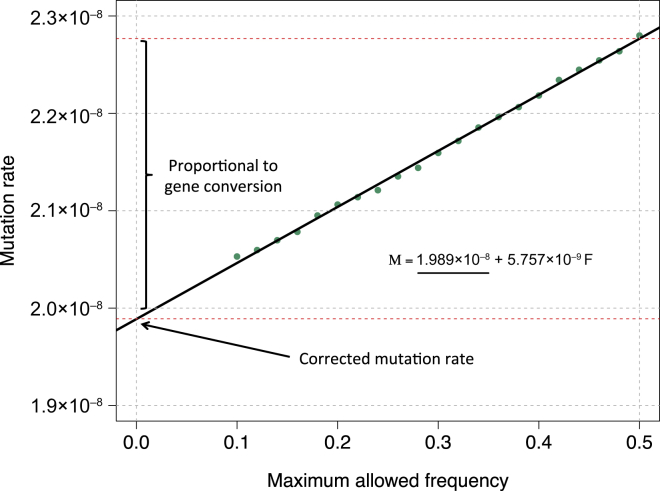
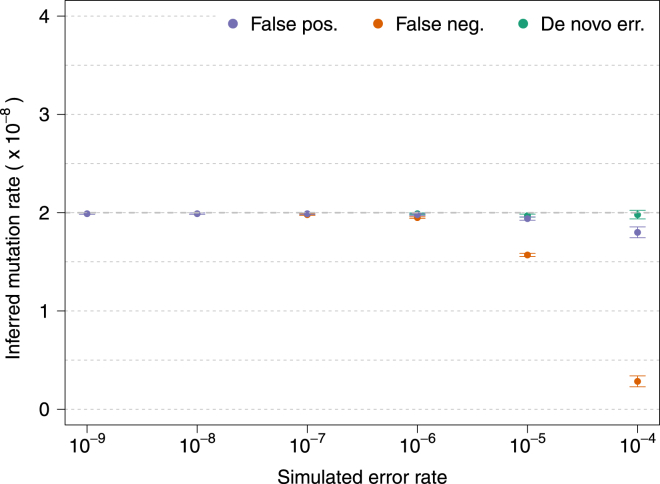
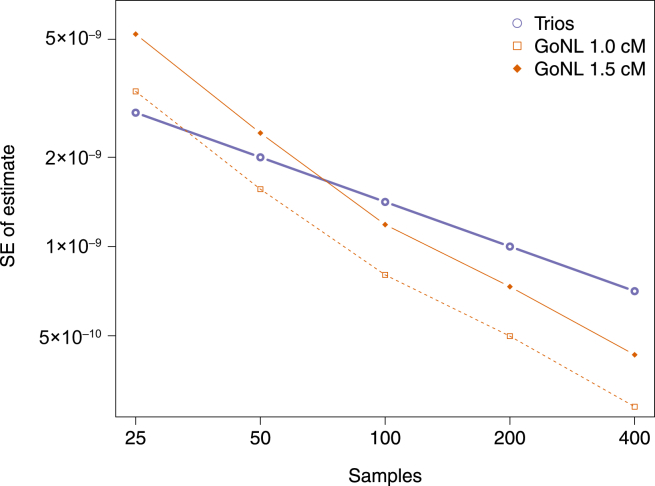
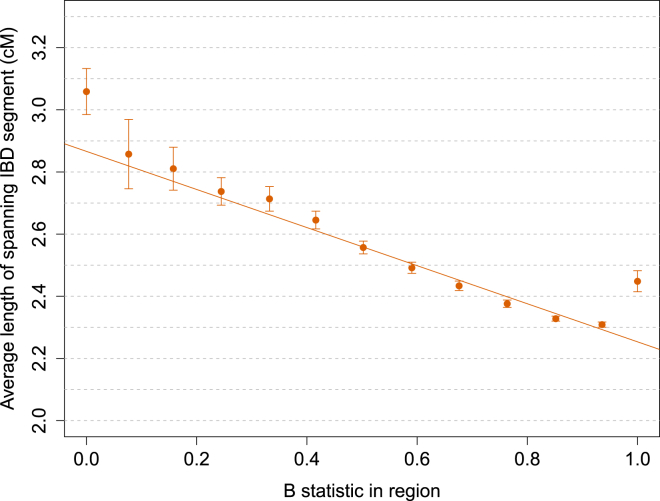
The rate at which human genomes mutate is a central biological parameter that has many implications for our ability to understand demographic and evolutionary phenomena. We present a method for inferring mutation and gene-conversion rates by using the number of sequence differences observed in identical-by-descent (IBD) segments together with a reconstructed model of recent population-size history. This approach is robust to, and can quantify, the presence of substantial genotyping error, as validated in coalescent simulations. We applied the method to 498 trio-phased sequenced Dutch individuals and inferred a point mutation rate of 1.66 × 10(-8) per base per generation and a rate of 1.26 × 10(-9) for <20 bp indels. By quantifying how estimates varied as a function of allele frequency, we inferred the probability that a site is involved in non-crossover gene conversion as 5.99 × 10(-6). We found that recombination does not have observable mutagenic effects after gene conversion is accounted for and that local gene-conversion rates reflect recombination rates. We detected a strong enrichment of recent deleterious variation among mismatching variants found within IBD regions and observed summary statistics of local sharing of IBD segments to closely match previously proposed metrics of background selection; however, we found no significant effects of selection on our mutation-rate estimates. We detected no evidence of strong variation of mutation rates in a number of genomic annotations obtained from several recent studies. Our analysis suggests that a mutation-rate estimate higher than that reported by recent pedigree-based studies should be adopted in the context of DNA-based demographic reconstruction.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
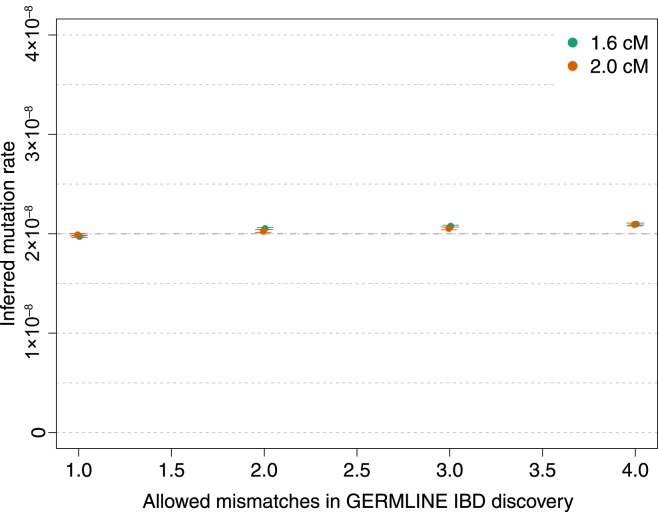
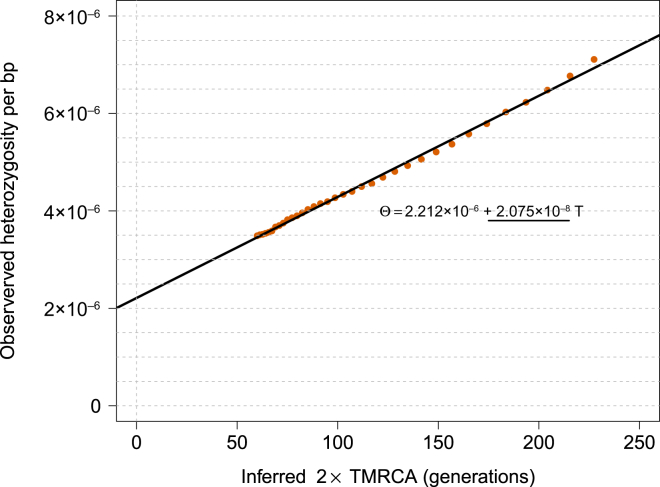
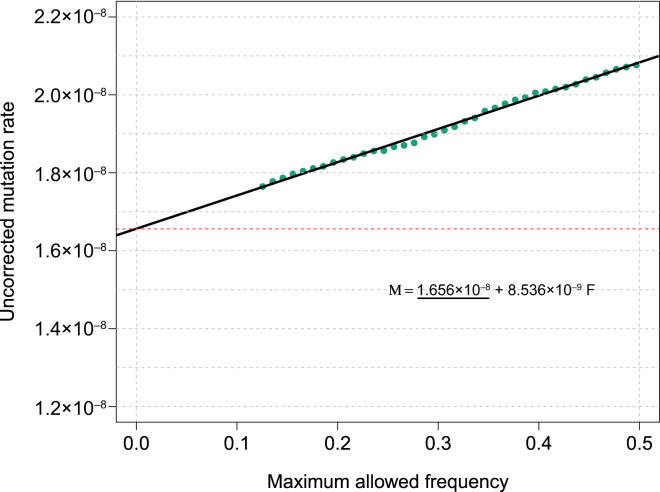
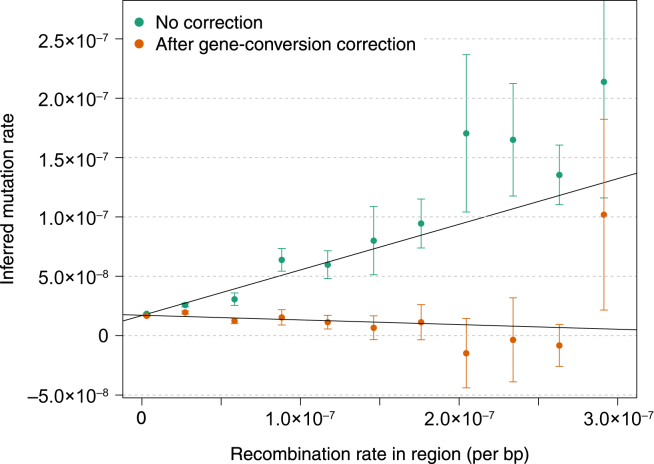
References
-
- Crow J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000;1:40–47. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
