

A manganese catalyst for highly reactive yet chemoselective intramolecular C(sp(3))-H amination
- PMID: 26587714
- PMCID: PMC4836951
- DOI: 10.1038/nchem.2366
A manganese catalyst for highly reactive yet chemoselective intramolecular C(sp(3))-H amination
Abstract
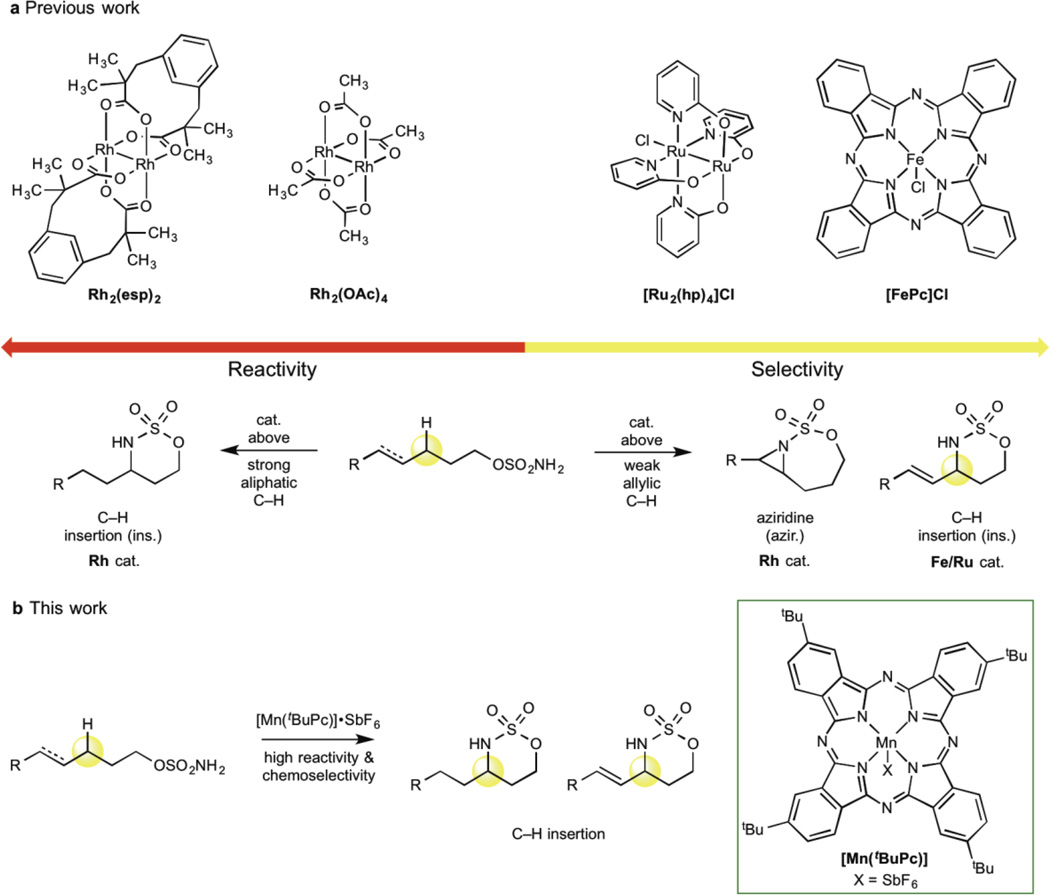
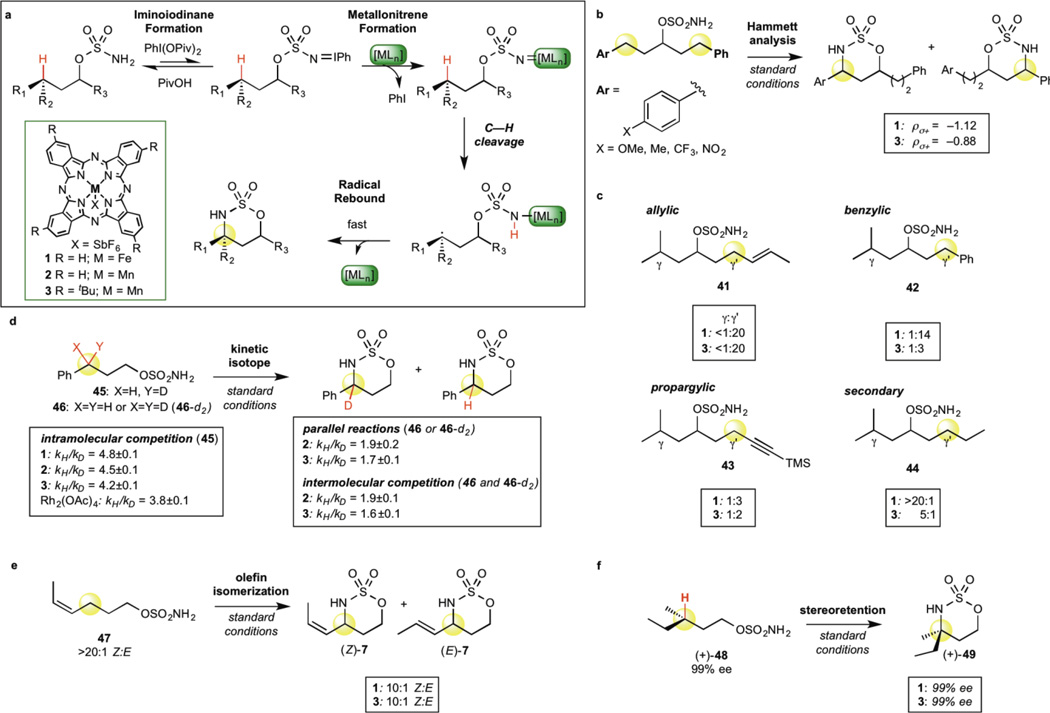
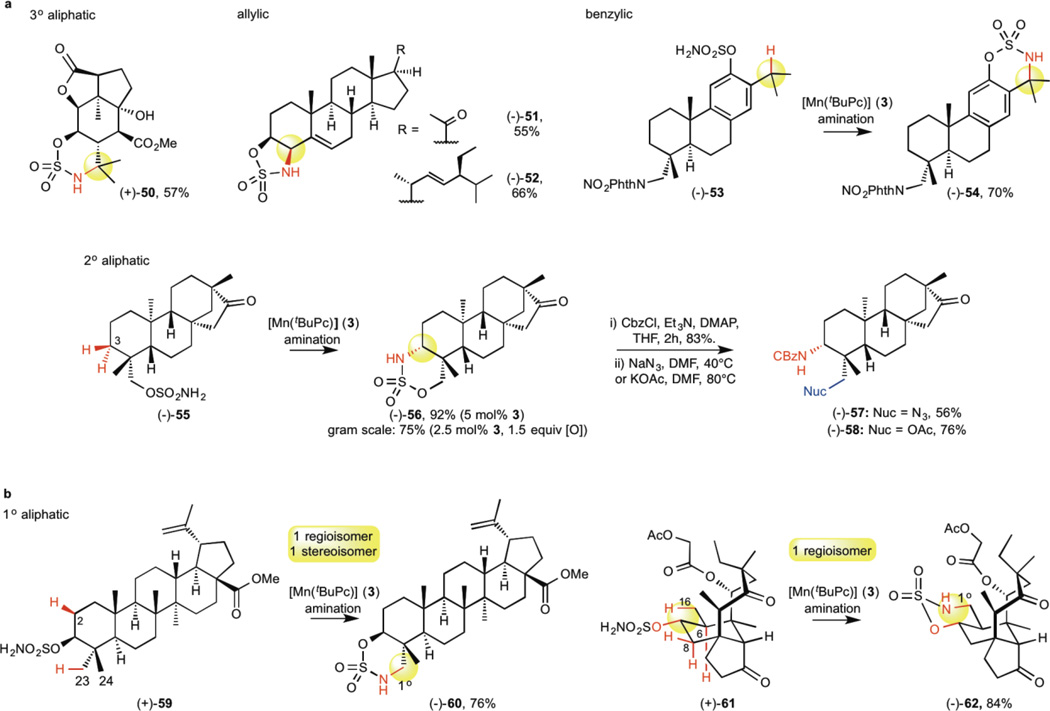
C-H bond oxidation reactions underscore the existing paradigm wherein high reactivity and high selectivity are inversely correlated. The development of catalysts capable of oxidizing strong aliphatic C(sp(3))-H bonds while displaying chemoselectivity (that is, tolerance of more oxidizable functionality) remains an unsolved problem. Here, we describe a catalyst, manganese tert-butylphthalocyanine [Mn((t)BuPc)], that is an outlier to the reactivity-selectivity paradigm. It is unique in its capacity to functionalize all types of C(sp(3))-H bond intramolecularly, while displaying excellent chemoselectivity in the presence of π functionality. Mechanistic studies indicate that [Mn((t)BuPc)] transfers bound nitrenes to C(sp(3))-H bonds via a pathway that lies between concerted C-H insertion, observed with reactive noble metals such as rhodium, and stepwise radical C-H abstraction/rebound, as observed with chemoselective base metals such as iron. Rather than achieving a blending of effects, [Mn((t)BuPc)] aminates even 1° aliphatic and propargylic C-H bonds, demonstrating reactivity and selectivity unusual for previously known catalysts.
Figures
References
-
- White MC. Adding aliphatic C—H bond oxidations to synthesis. Science. 2012;335:807–809. - PubMed
-
- Que L., Jr The road to non-heme oxoferryls and beyond. Acc. Chem. Res. 2007;40:493–500. - PubMed
-
- Nam W, Lee Y, Fukuzumi S. Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron(IV)-oxo complexes. Acc. Chem. Res. 2014;47:1146–1154. - PubMed
-
- Chen MS, White MCA. predictable selective aliphatic C—H oxidation reaction for complex molecule synthesis. Science. 2007;318:783–787. - PubMed
Publication types
MeSH terms
Substances
Associated data
- PubChem-Substance/252275717
- PubChem-Substance/252275718
- PubChem-Substance/252275719
- PubChem-Substance/252275720
- PubChem-Substance/252275721
- PubChem-Substance/252275722
- PubChem-Substance/252275723
- PubChem-Substance/252275724
- PubChem-Substance/252275725
- PubChem-Substance/252275726
- PubChem-Substance/252275727
- PubChem-Substance/252275728
- PubChem-Substance/252275729
- PubChem-Substance/252275730
- PubChem-Substance/252275731
- PubChem-Substance/252275732
- PubChem-Substance/252275733
- PubChem-Substance/252275734
- PubChem-Substance/252275735
- PubChem-Substance/252275736
- PubChem-Substance/252275737
- PubChem-Substance/252275738
- PubChem-Substance/252275739
- PubChem-Substance/252275740
- PubChem-Substance/252275741
- PubChem-Substance/252275742
- PubChem-Substance/252275743
- PubChem-Substance/252275744
- PubChem-Substance/252275745
- PubChem-Substance/252275746
- PubChem-Substance/252275747
- PubChem-Substance/252275748
- PubChem-Substance/252297299
- PubChem-Substance/252297300
- PubChem-Substance/252297301
- PubChem-Substance/252297302
- PubChem-Substance/252297303
- PubChem-Substance/252297304
- PubChem-Substance/252297305
- PubChem-Substance/252297306
- PubChem-Substance/252297307
- PubChem-Substance/252297308
- PubChem-Substance/252297309
- PubChem-Substance/252297310
- PubChem-Substance/252297311
- PubChem-Substance/252297312
- PubChem-Substance/252297313
- PubChem-Substance/252297314
- PubChem-Substance/252297315
- PubChem-Substance/252297316
- PubChem-Substance/252297317
- PubChem-Substance/252297318
- PubChem-Substance/252297319
- PubChem-Substance/252297320
- PubChem-Substance/252297321
- PubChem-Substance/252297322
- PubChem-Substance/252297323
- PubChem-Substance/252297324
- PubChem-Substance/252297325
- PubChem-Substance/252297326
- PubChem-Substance/252297327
- PubChem-Substance/252297328
- PubChem-Substance/252297329
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
