

Mitochondrial Quality Control as a Therapeutic Target
- PMID: 26589414
- PMCID: PMC11060432
- DOI: 10.1124/pr.115.011502
Mitochondrial Quality Control as a Therapeutic Target
Abstract
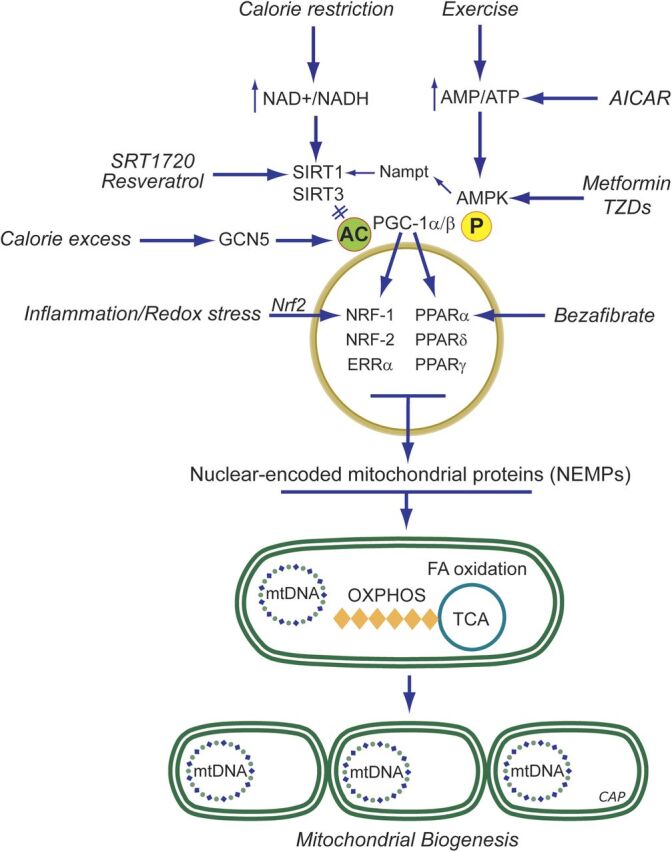
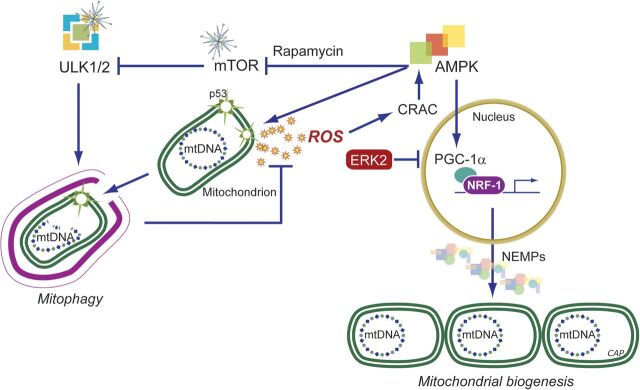
In addition to oxidative phosphorylation (OXPHOS), mitochondria perform other functions such as heme biosynthesis and oxygen sensing and mediate calcium homeostasis, cell growth, and cell death. They participate in cell communication and regulation of inflammation and are important considerations in aging, drug toxicity, and pathogenesis. The cell's capacity to maintain its mitochondria involves intramitochondrial processes, such as heme and protein turnover, and those involving entire organelles, such as fusion, fission, selective mitochondrial macroautophagy (mitophagy), and mitochondrial biogenesis. The integration of these processes exemplifies mitochondrial quality control (QC), which is also important in cellular disorders ranging from primary mitochondrial genetic diseases to those that involve mitochondria secondarily, such as neurodegenerative, cardiovascular, inflammatory, and metabolic syndromes. Consequently, mitochondrial biology represents a potentially useful, but relatively unexploited area of therapeutic innovation. In patients with genetic OXPHOS disorders, the largest group of inborn errors of metabolism, effective therapies, apart from symptomatic and nutritional measures, are largely lacking. Moreover, the genetic and biochemical heterogeneity of these states is remarkably similar to those of certain acquired diseases characterized by metabolic and oxidative stress and displaying wide variability. This biologic variability reflects cell-specific and repair processes that complicate rational pharmacological approaches to both primary and secondary mitochondrial disorders. However, emerging concepts of mitochondrial turnover and dynamics along with new mitochondrial disease models are providing opportunities to develop and evaluate mitochondrial QC-based therapies. The goals of such therapies extend beyond amelioration of energy insufficiency and tissue loss and entail cell repair, cell replacement, and the prevention of fibrosis. This review summarizes current concepts of mitochondria as disease elements and outlines novel strategies to address mitochondrial dysfunction through the stimulation of mitochondrial biogenesis and quality control.
Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
References
-
- Alam J, Igarashi K, Immenschuh S, Shibahara S, Tyrrell RM. (2004) Regulation of heme oxygenase-1 gene transcription: recent advances and highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid Redox Signal 6:924–933. - PubMed
-
- Archer DF. (2010) Tissue-selective estrogen complexes: a promising option for the comprehensive management of menopausal symptoms. Drugs Aging 27:533–544. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
