

M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia
- PMID: 26590347
- PMCID: PMC4864040
- DOI: 10.1016/j.neuron.2015.10.039
M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia
Erratum in
-
M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia.Neuron. 2016 Jun 1;90(5):1139. doi: 10.1016/j.neuron.2016.05.017. Neuron. 2016. PMID: 27253449 Free PMC article. No abstract available.
Abstract
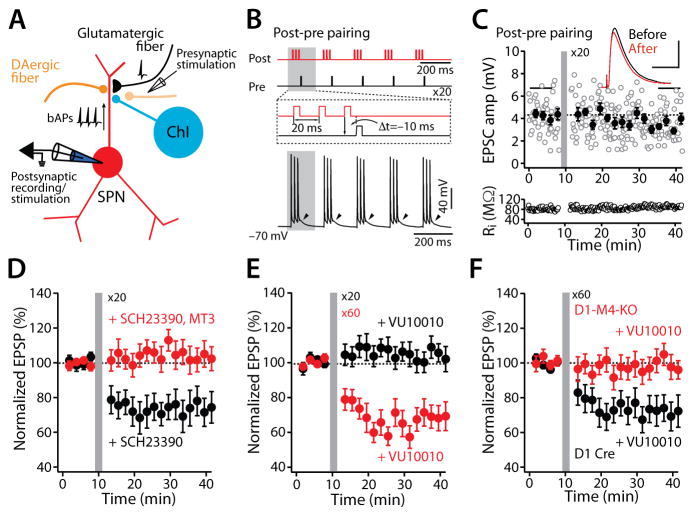
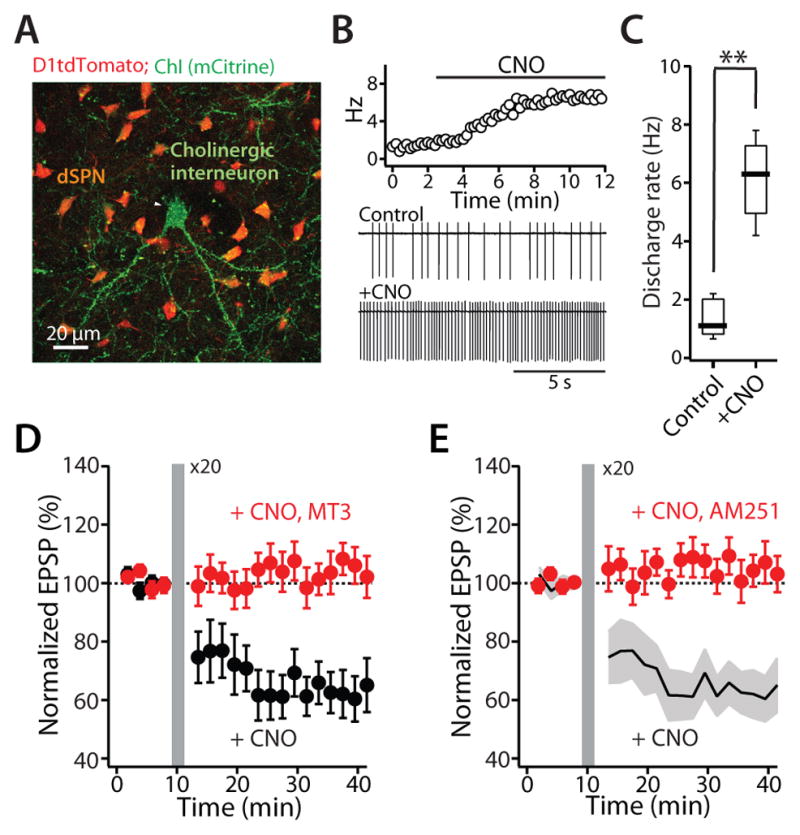
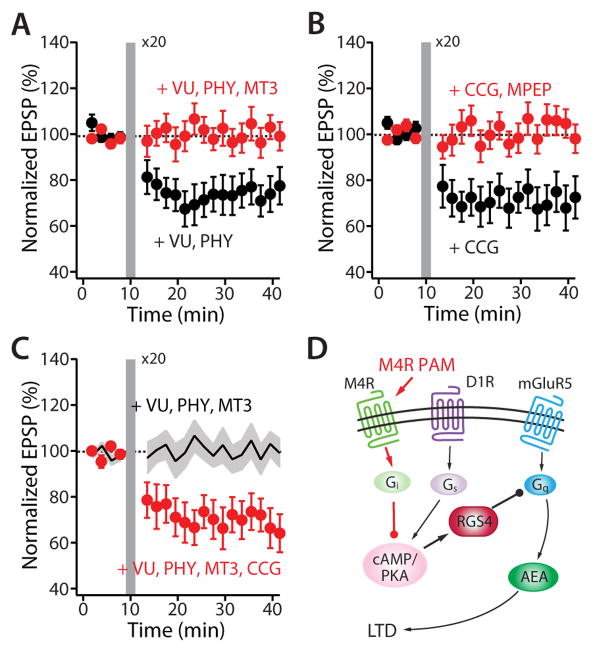
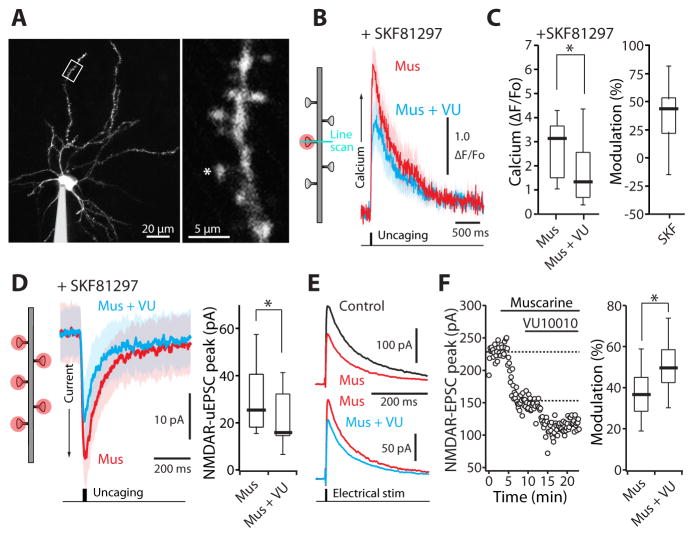
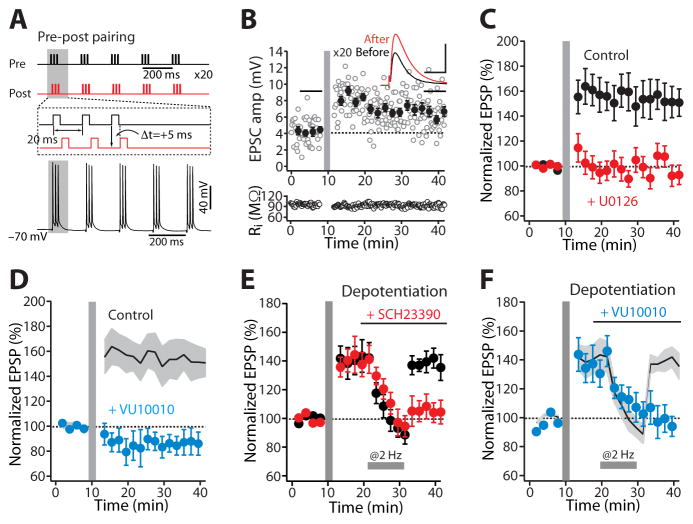
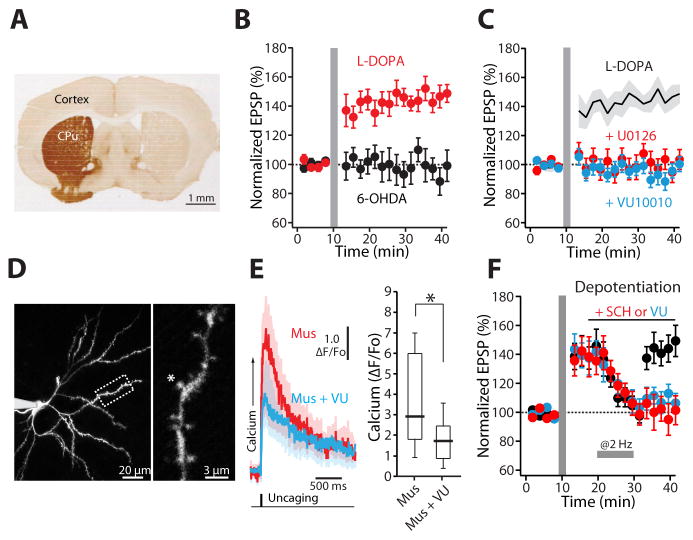
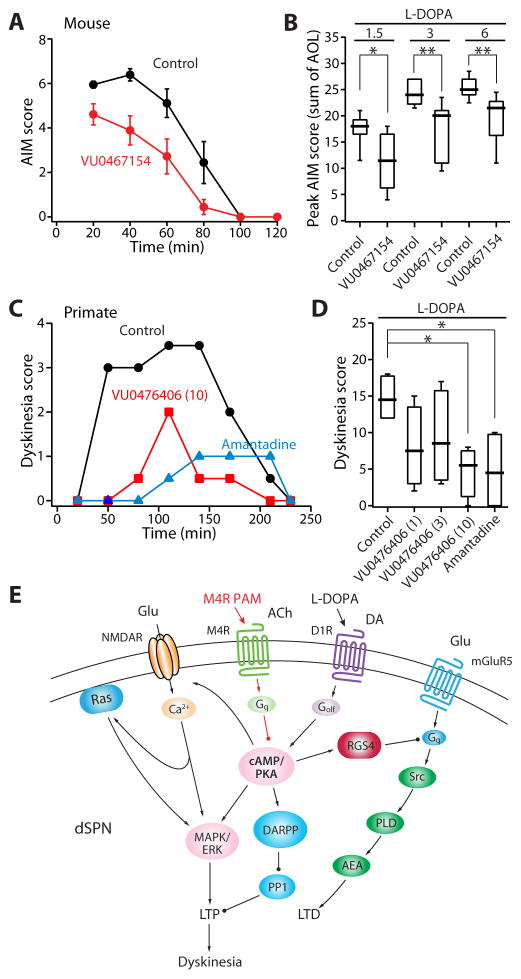
A balanced interaction between dopaminergic and cholinergic signaling in the striatum is critical to goal-directed behavior. But how this interaction modulates corticostriatal synaptic plasticity underlying learned actions remains unclear--particularly in direct-pathway spiny projection neurons (dSPNs). Our studies show that in dSPNs, endogenous cholinergic signaling through M4 muscarinic receptors (M4Rs) promoted long-term depression of corticostriatal glutamatergic synapses, by suppressing regulator of G protein signaling type 4 (RGS4) activity, and blocked D1 dopamine receptor dependent long-term potentiation (LTP). Furthermore, in a mouse model of L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesia (LID) in Parkinson's disease (PD), boosting M4R signaling with positive allosteric modulator (PAM) blocked aberrant LTP in dSPNs, enabled LTP reversal, and attenuated dyskinetic behaviors. An M4R PAM also was effective in a primate LID model. Taken together, these studies identify an important signaling pathway controlling striatal synaptic plasticity and point to a novel pharmacological strategy for alleviating LID in PD patients.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Bridging the Gap: Muscarinic M4 Receptors Promote Striatal Plasticity in Health and Disease.Neuron. 2015 Nov 18;88(4):621-3. doi: 10.1016/j.neuron.2015.11.007. Neuron. 2015. PMID: 26590337
References
-
- Bezard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med. 2003;9:762–767. - PubMed
-
- Blazer LL, Storaska AJ, Jutkiewicz EM, Turner EM, Calcagno M, Wade SM, Wang Q, Huang XP, Traynor JR, Husbands SM, et al. Selectivity and anti-Parkinson’s potential of thiadiazolidinone RGS4 inhibitors. ACS Chem Neurosci. 2015;6:911–919. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
