

RNA mis-splicing in disease
- PMID: 26593421
- PMCID: PMC5993438
- DOI: 10.1038/nrg.2015.3
RNA mis-splicing in disease
Abstract
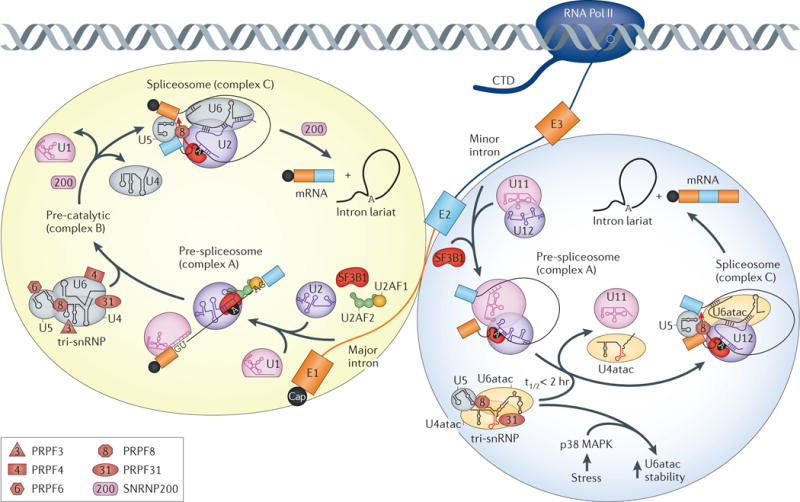
The human transcriptome is composed of a vast RNA population that undergoes further diversification by splicing. Detecting specific splice sites in this large sequence pool is the responsibility of the major and minor spliceosomes in collaboration with numerous splicing factors. This complexity makes splicing susceptible to sequence polymorphisms and deleterious mutations. Indeed, RNA mis-splicing underlies a growing number of human diseases with substantial societal consequences. Here, we provide an overview of RNA splicing mechanisms followed by a discussion of disease-associated errors, with an emphasis on recently described mutations that have provided new insights into splicing regulation. We also discuss emerging strategies for splicing-modulating therapy.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Wilhelm M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. - PubMed
-
- Treutlein B, Gokce O, Quake SR, Sudhof TC. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc Natl Acad Sci USA. 2014;111:E1291–E1299. Long-read sequencing of full-length neurexin mRNAs from pre-frontal cortex is performed to determine the extent of alternative splicing and provide evidence for thousands of neurexin isoforms. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
