

New Insights into the Pros and Cons of the Clinical Use of Vitamin K Antagonists (VKAs) Versus Direct Oral Anticoagulants (DOACs)
- PMID: 26593943
- PMCID: PMC4663607
- DOI: 10.3390/nu7115479
New Insights into the Pros and Cons of the Clinical Use of Vitamin K Antagonists (VKAs) Versus Direct Oral Anticoagulants (DOACs)
Abstract
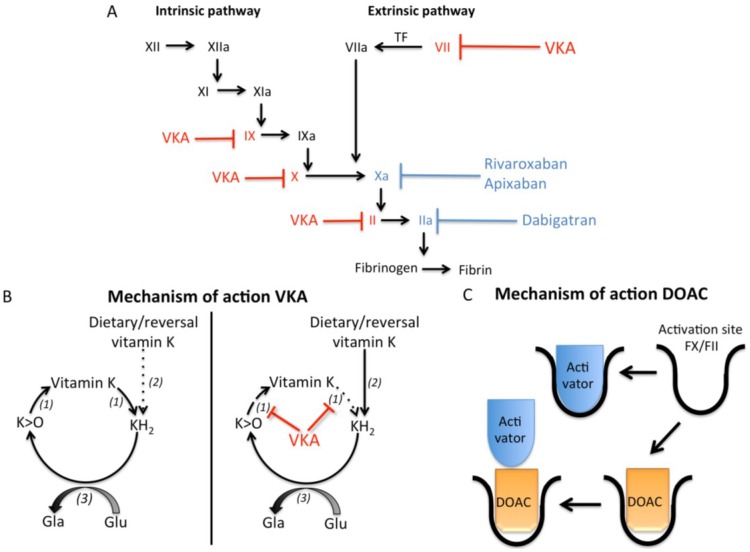
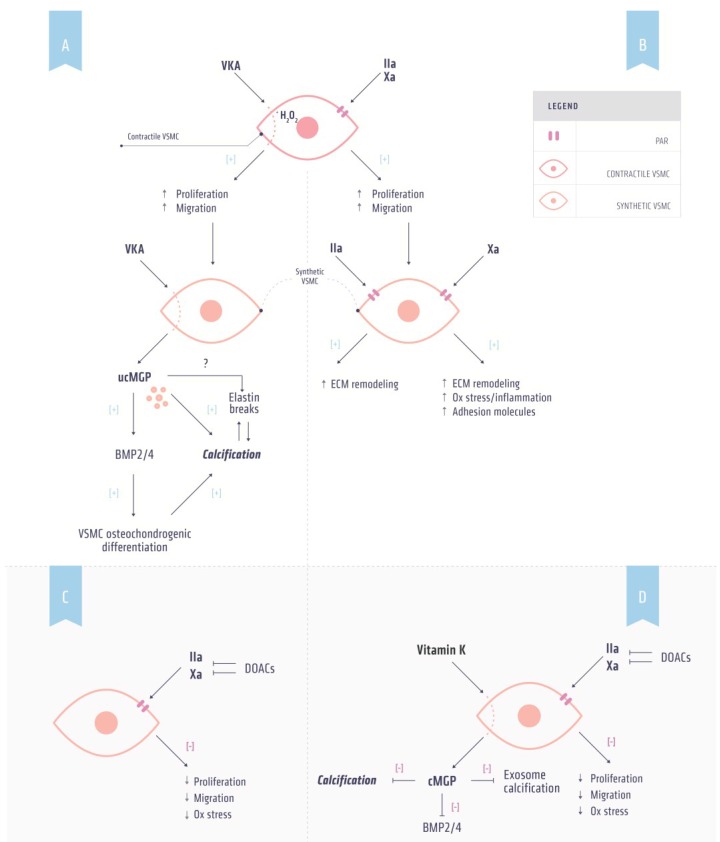
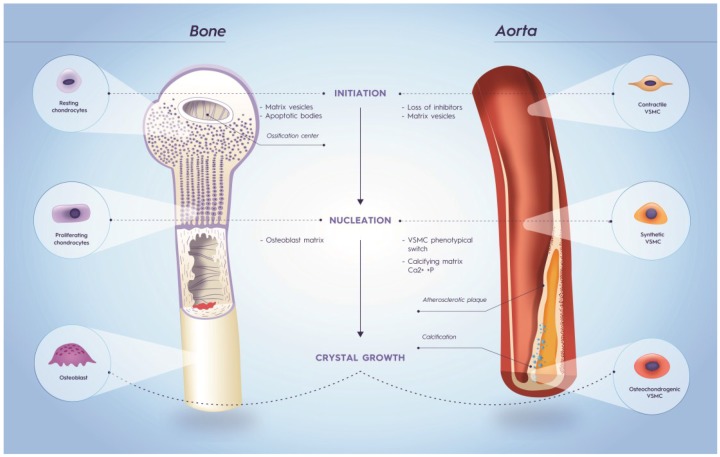
Vitamin K-antagonists (VKA) are the most widely used anticoagulant drugs to treat patients at risk of arterial and venous thrombosis for the past 50 years. Due to unfavorable pharmacokinetics VKA have a small therapeutic window, require frequent monitoring, and are susceptible to drug and nutritional interactions. Additionally, the effect of VKA is not limited to coagulation, but affects all vitamin K-dependent proteins. As a consequence, VKA have detrimental side effects by enhancing medial and intimal calcification. These limitations stimulated the development of alternative anticoagulant drugs, resulting in direct oral anticoagulant (DOAC) drugs, which specifically target coagulation factor Xa and thrombin. DOACs also display non-hemostatic vascular effects via protease-activated receptors (PARs). As atherosclerosis is characterized by a hypercoagulable state indicating the involvement of activated coagulation factors in the genesis of atherosclerosis, anticoagulation could have beneficial effects on atherosclerosis. Additionally, accumulating evidence demonstrates vascular benefit from high vitamin K intake. This review gives an update on oral anticoagulant treatment on the vasculature with a special focus on calcification and vitamin K interaction.
Keywords: DOACs; coumarin; oral anticoagulants; vascular calcification; vitamin K.
Figures
References
-
- Mann K.G. Biochemistry and physiology of blood coagulation. Thromb. Haemost. 1999;82:165–174. - PubMed
-
- Badimon L. Atherosclerosis and thrombosis: Lessons from animal models. Thromb. Haemost. 2001;86:356–365. - PubMed
-
- Fuster V., Stein B., Ambrose J.A., Badimon L., Badimon J.J., Chesebro J.H. Atherosclerotic plaque rupture and thrombosis. Evolving concepts. Circulation. 1990;82:II47–II59. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
