

Traumatic Brain Injury and Peripheral Immune Suppression: Primer and Prospectus
- PMID: 26594196
- PMCID: PMC4633482
- DOI: 10.3389/fneur.2015.00235
Traumatic Brain Injury and Peripheral Immune Suppression: Primer and Prospectus
Abstract
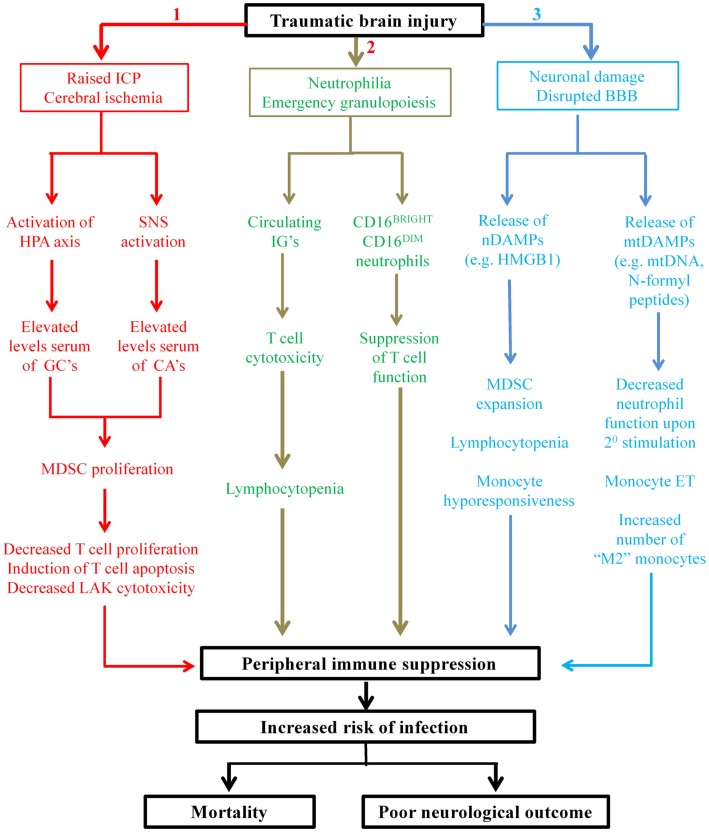
Nosocomial infections are a common occurrence in patients following traumatic brain injury (TBI) and are associated with an increased risk of mortality, longer length of hospital stay, and poor neurological outcome. Systemic immune suppression arising as a direct result of injury to the central nervous system (CNS) is considered to be primarily responsible for this increased incidence of infection, a view strengthened by recent studies that have reported novel changes in the composition and function of the innate and adaptive arms of the immune system post-TBI. However, our knowledge of the mechanisms that underlie TBI-induced immune suppression is equivocal at best. Here, after summarizing our current understanding of the impact of TBI on peripheral immunity and discussing CNS-mediated regulation of immune function, we propose roles for a series of novel mechanisms in driving the immune suppression that is observed post-TBI. These mechanisms, which have never been considered before in the context of TBI-induced immune paresis, include the CNS-driven emergence into the circulation of myeloid-derived suppressor cells and suppressive neutrophil subsets, and the release from injured tissue of nuclear and mitochondria-derived damage associated molecular patterns. Moreover, in an effort to further our understanding of the mechanisms that underlie TBI-induced changes in immunity, we pose throughout the review a series of questions, which if answered would address a number of key issues, such as establishing whether manipulating peripheral immune function has potential as a future therapeutic strategy by which to treat and/or prevent infections in the hospitalized TBI patient.
Keywords: immune suppression; immune system; infection; traumatic brain injury.
Figures
References
-
- Menon DK, Schwab K, Wright DW, Maas AI, Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health . Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil (2010) 91:1637–40.10.1016/j.apmr.2010.05.017 - DOI - PubMed
-
- Health and Social Care Information Centre. Hospital Episode Statistics, Admitted Patient Care, England 2012-13: Diagnosis (2015). Available from: http://www.hscic.gov.uk/catalogue/PUB16719
-
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; (2010).
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources