

Multiplexed analysis of chromosome conformation at vastly improved sensitivity
- PMID: 26595209
- PMCID: PMC4724891
- DOI: 10.1038/nmeth.3664
Multiplexed analysis of chromosome conformation at vastly improved sensitivity
Abstract
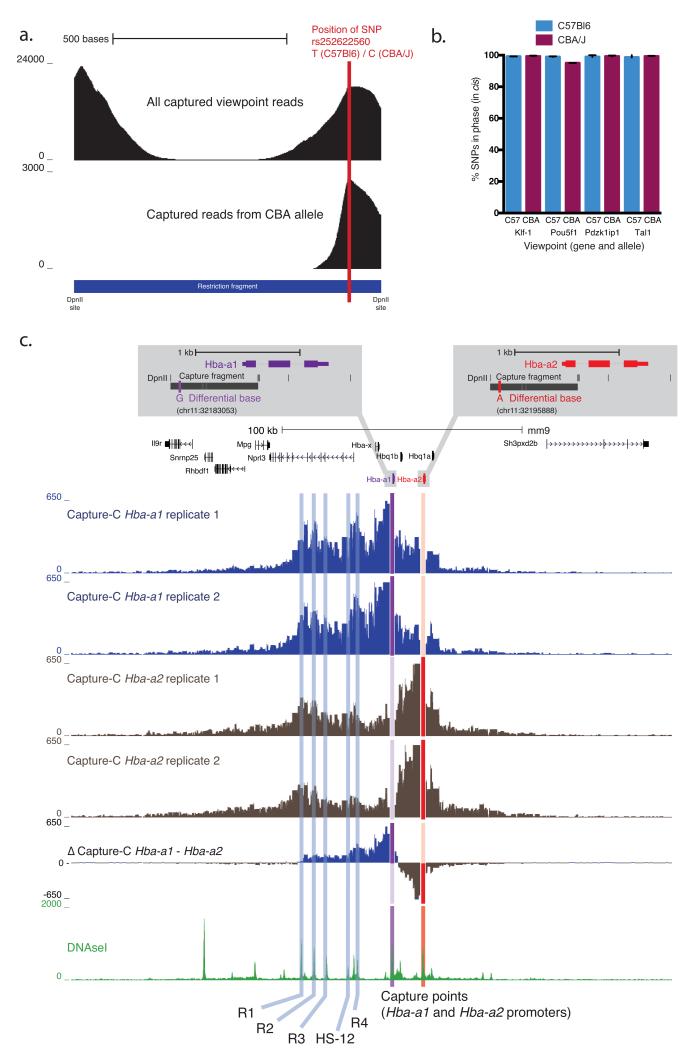
Methods for analyzing chromosome conformation in mammalian cells are either low resolution or low throughput and are technically challenging. In next-generation (NG) Capture-C, we have redesigned the Capture-C method to achieve unprecedented levels of sensitivity and reproducibility. NG Capture-C can be used to analyze many genetic loci and samples simultaneously. High-resolution data can be produced with as few as 100,000 cells, and single-nucleotide polymorphisms can be used to generate allele-specific tracks. The method is straightforward to perform and should greatly facilitate the investigation of many questions related to gene regulation as well as the functional dissection of traits examined in genome-wide association studies.
Figures



Similar articles
-
High-throughput Identification of Gene Regulatory Sequences Using Next-generation Sequencing of Circular Chromosome Conformation Capture (4C-seq).J Vis Exp. 2018 Oct 5;(140):58030. doi: 10.3791/58030. J Vis Exp. 2018. PMID: 30346381 Free PMC article.
-
Detection of Short-Range DNA Interactions in Mammalian Cells Using High-Resolution Circular Chromosome Conformation Capture Coupled to Deep Sequencing.Methods Mol Biol. 2015;1334:245-59. doi: 10.1007/978-1-4939-2877-4_16. Methods Mol Biol. 2015. PMID: 26404155
-
Assessment of 3D Interactions Between Promoters and Distal Regulatory Elements with Promoter Capture Hi-C (PCHi-C).Methods Mol Biol. 2021;2351:229-248. doi: 10.1007/978-1-0716-1597-3_13. Methods Mol Biol. 2021. PMID: 34382193
-
Novel insights into chromosomal conformations in cancer.Mol Cancer. 2017 Nov 17;16(1):173. doi: 10.1186/s12943-017-0741-5. Mol Cancer. 2017. PMID: 29149895 Free PMC article. Review.
-
Understanding and utilizing crop genome diversity via high-resolution genotyping.Plant Biotechnol J. 2016 Apr;14(4):1086-94. doi: 10.1111/pbi.12456. Epub 2015 Aug 19. Plant Biotechnol J. 2016. PMID: 27003869 Free PMC article. Review.
Cited by
-
The macro and micro of chromosome conformation capture.Wiley Interdiscip Rev Dev Biol. 2021 Nov;10(6):e395. doi: 10.1002/wdev.395. Epub 2020 Sep 28. Wiley Interdiscip Rev Dev Biol. 2021. PMID: 32987449 Free PMC article. Review.
-
HiChIP: efficient and sensitive analysis of protein-directed genome architecture.Nat Methods. 2016 Nov;13(11):919-922. doi: 10.1038/nmeth.3999. Epub 2016 Sep 19. Nat Methods. 2016. PMID: 27643841 Free PMC article.
-
A modified protocol of Capture-C allows affordable and flexible high-resolution promoter interactome analysis.Sci Rep. 2020 Sep 23;10(1):15491. doi: 10.1038/s41598-020-72496-4. Sci Rep. 2020. PMID: 32968144 Free PMC article.
-
A Conserved Noncoding Locus Regulates Random Monoallelic Xist Expression across a Topological Boundary.Mol Cell. 2020 Jan 16;77(2):352-367.e8. doi: 10.1016/j.molcel.2019.10.030. Epub 2019 Nov 20. Mol Cell. 2020. PMID: 31759823 Free PMC article.
-
Essential role of Cp190 in physical and regulatory boundary formation.Sci Adv. 2022 May 13;8(19):eabl8834. doi: 10.1126/sciadv.abl8834. Epub 2022 May 13. Sci Adv. 2022. PMID: 35559678 Free PMC article.
References
-
- Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–7. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
