

Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice
- PMID: 26595809
- PMCID: PMC4701549
- DOI: 10.1172/JCI84669
Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice
Abstract
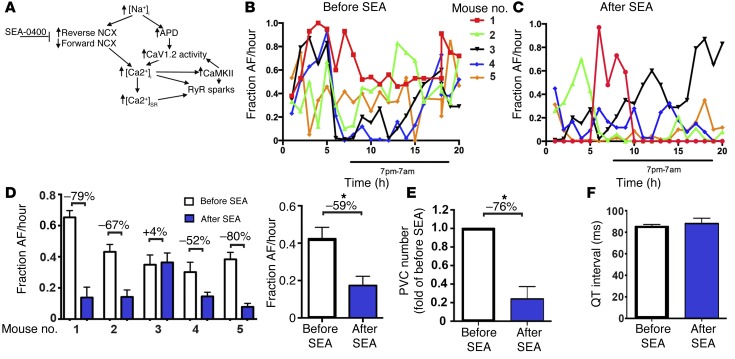
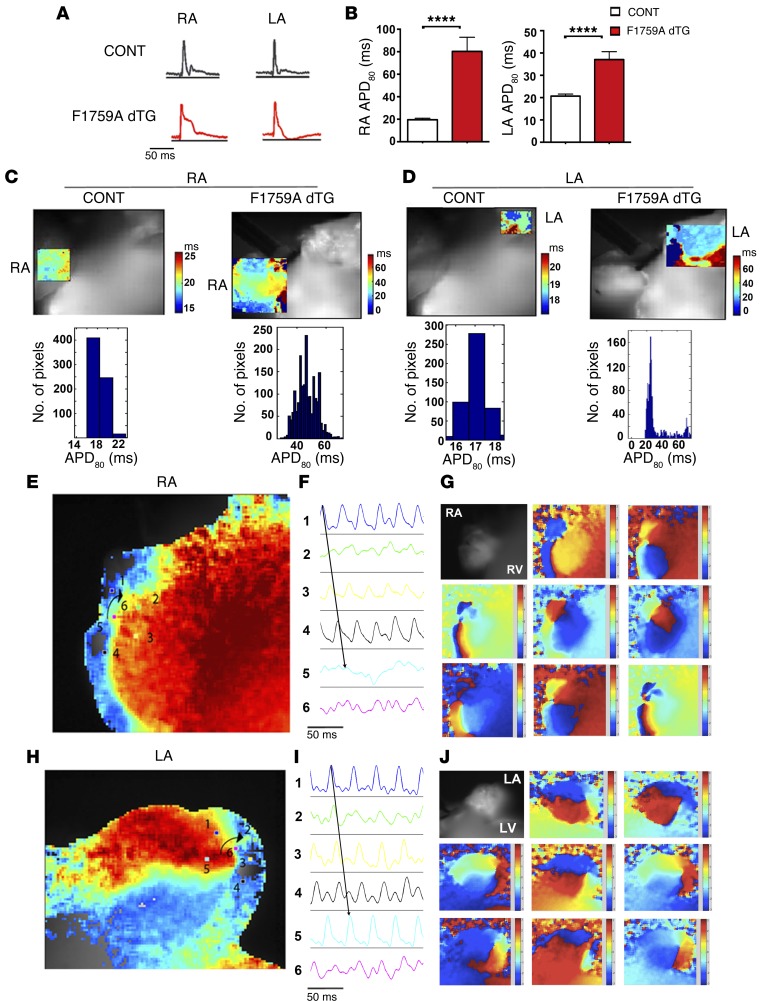
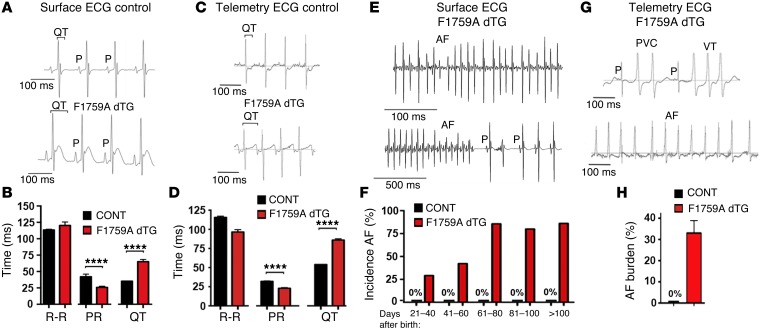
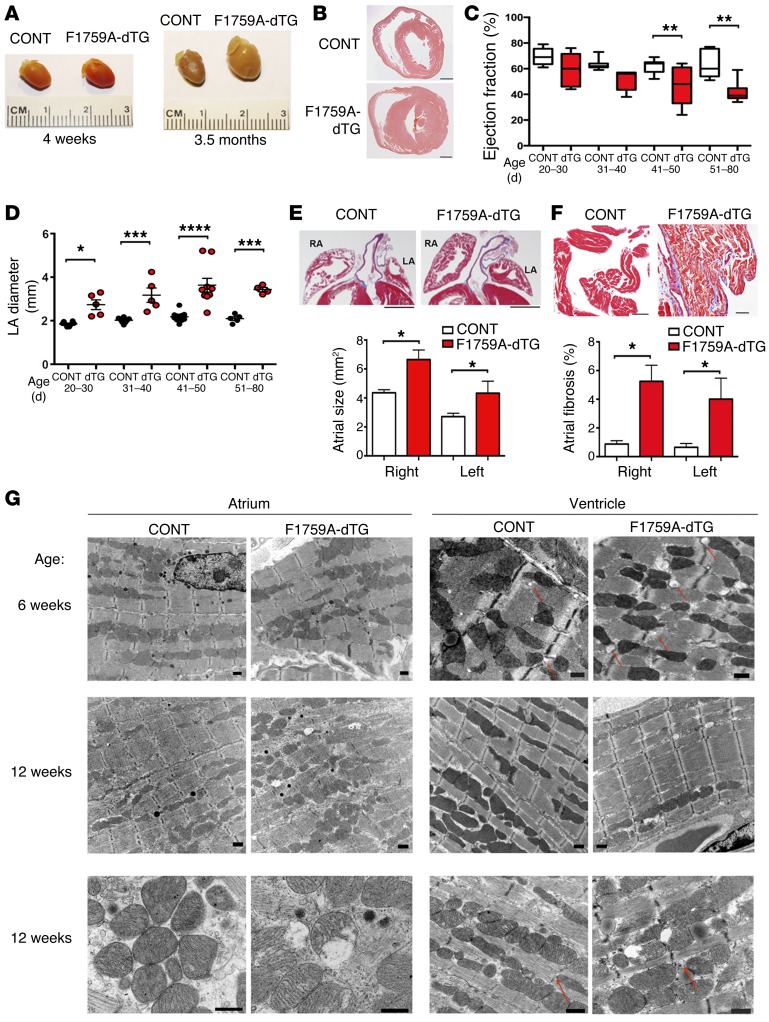
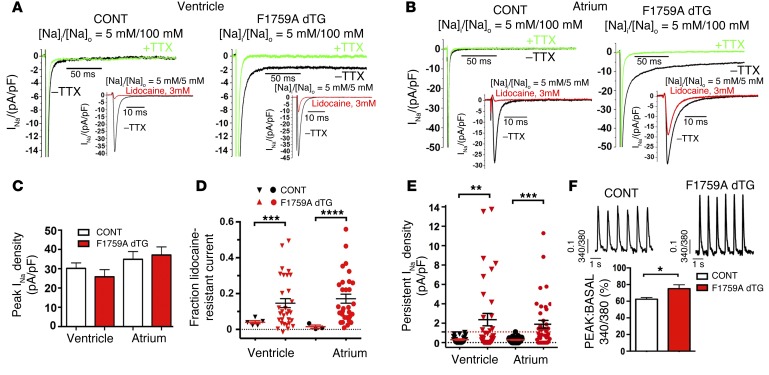
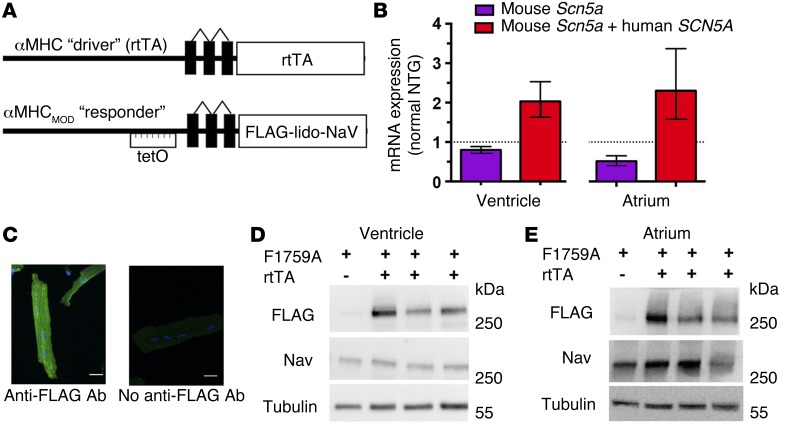
Increased sodium influx via incomplete inactivation of the major cardiac sodium channel Na(V)1.5 is correlated with an increased incidence of atrial fibrillation (AF) in humans. Here, we sought to determine whether increased sodium entry is sufficient to cause the structural and electrophysiological perturbations that are required to initiate and sustain AF. We used mice expressing a human Na(V)1.5 variant with a mutation in the anesthetic-binding site (F1759A-Na(V)1.5) and demonstrated that incomplete Na+ channel inactivation is sufficient to drive structural alterations, including atrial and ventricular enlargement, myofibril disarray, fibrosis and mitochondrial injury, and electrophysiological dysfunctions that together lead to spontaneous and prolonged episodes of AF in these mice. Using this model, we determined that the increase in a persistent sodium current causes heterogeneously prolonged action potential duration and rotors, as well as wave and wavelets in the atria, and thereby mimics mechanistic theories that have been proposed for AF in humans. Acute inhibition of the sodium-calcium exchanger, which targets the downstream effects of enhanced sodium entry, markedly reduced the burden of AF and ventricular arrhythmias in this model, suggesting a potential therapeutic approach for AF. Together, our results indicate that these mice will be important for assessing the cellular mechanisms and potential effectiveness of antiarrhythmic therapies.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
