

Nitrated fatty acids suppress angiotensin II-mediated fibrotic remodelling and atrial fibrillation
- PMID: 26598510
- PMCID: PMC5901131
- DOI: 10.1093/cvr/cvv254
Nitrated fatty acids suppress angiotensin II-mediated fibrotic remodelling and atrial fibrillation
Abstract
Aim: Atrial fibrosis, one of the most striking features in the pathology of atrial fibrillation (AF), is promoted by local and systemic inflammation. Electrophilic fatty acid nitroalkenes, endogenously generated by both metabolic and inflammatory reactions, are anti-inflammatory mediators that in synthetic form may be useful as drug candidates. Herein we investigate whether an exemplary nitro-fatty acid can limit atrial fibrosis and AF.
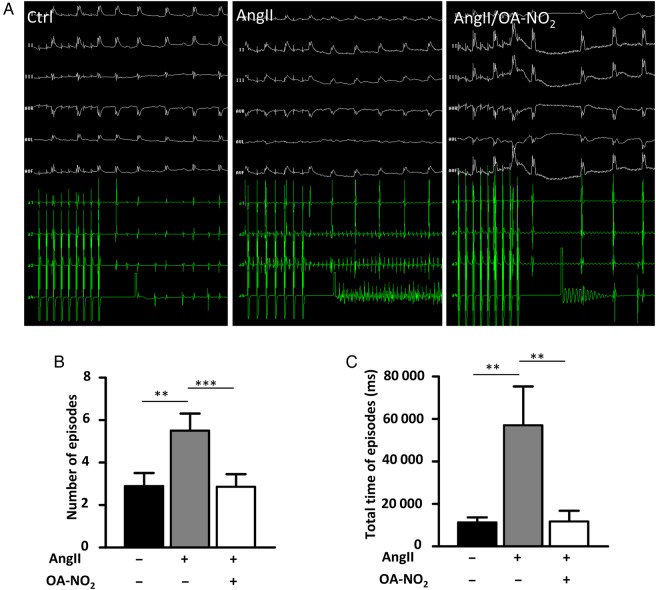
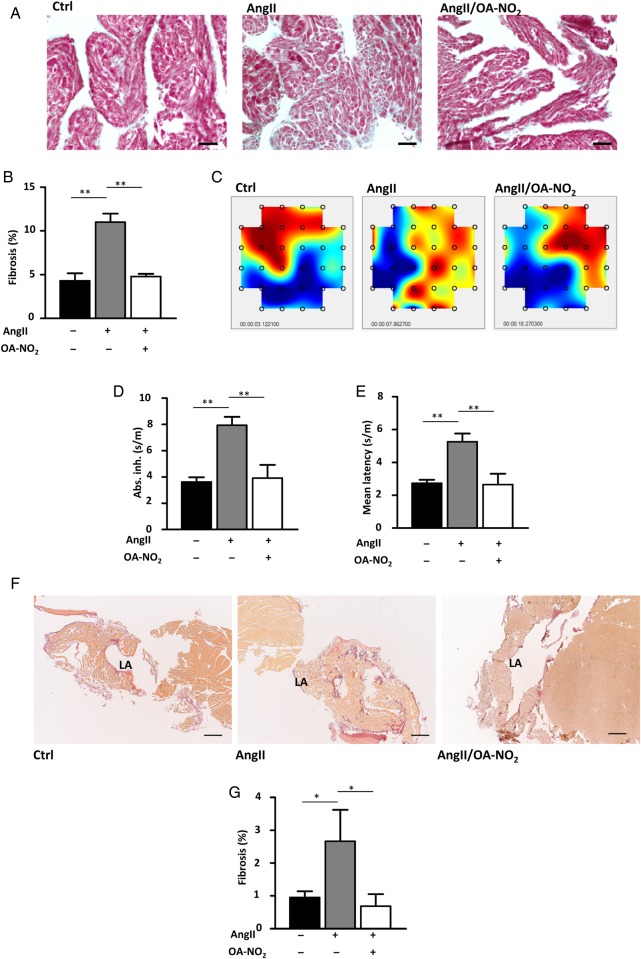
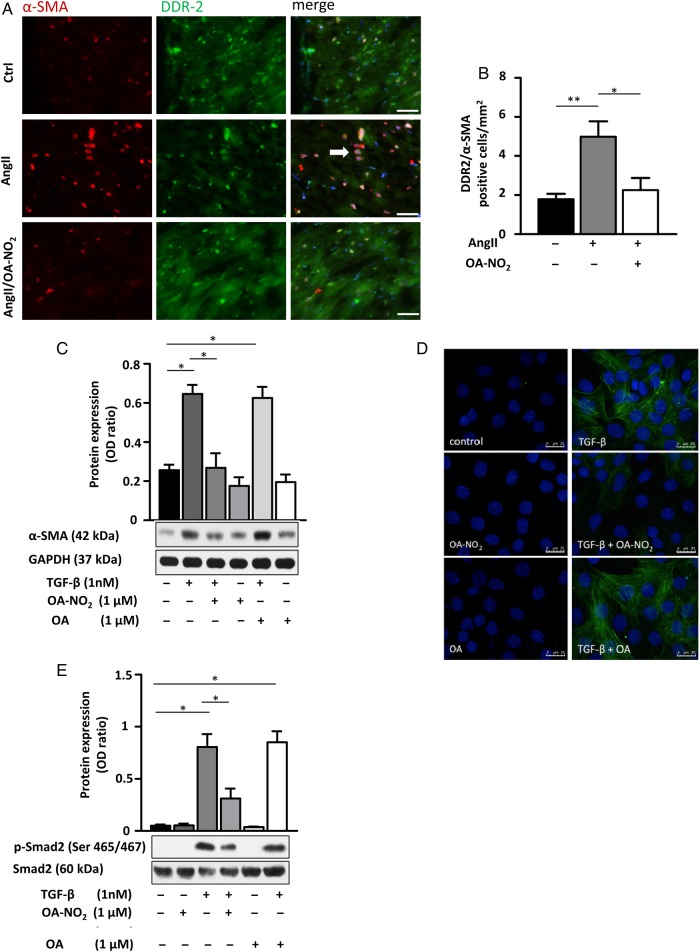
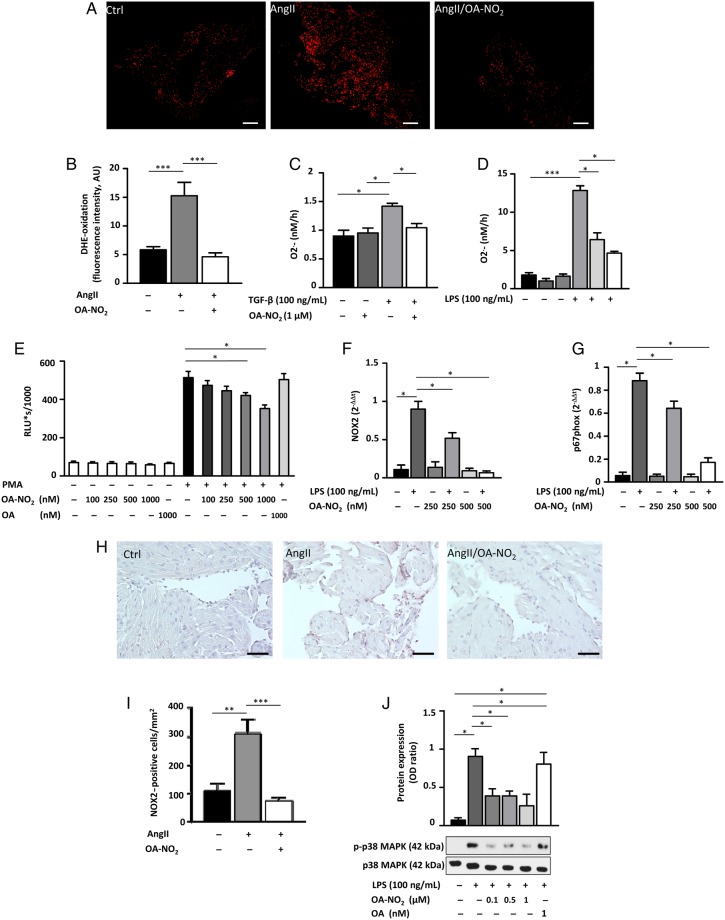
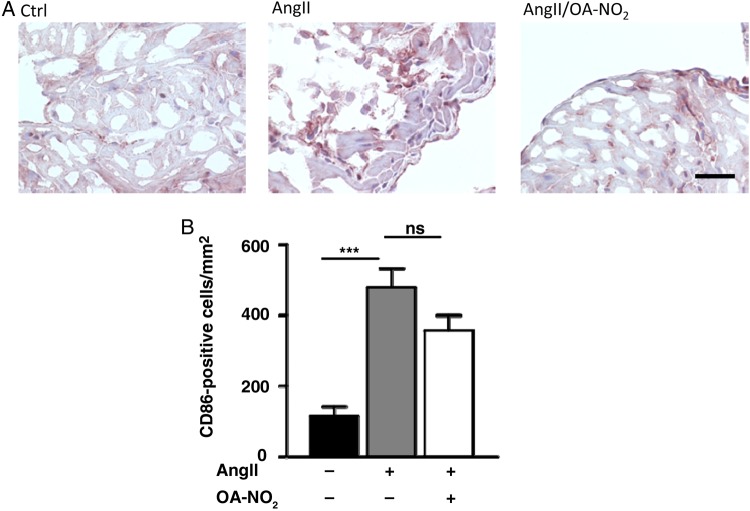
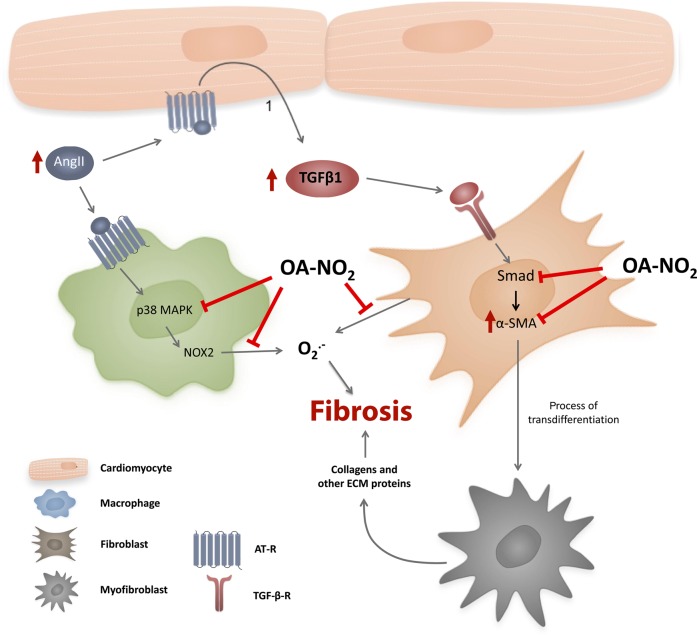
Methods and results: Wild-type C57BL6/J mice were treated for 2 weeks with angiotensin II (AngII) and vehicle or nitro-oleic acid (10-nitro-octadec-9-enoic acid, OA-NO2, 6 mg/kg body weight) via subcutaneous osmotic minipumps. OA-NO2 significantly inhibited atrial fibrosis and depressed vulnerability for AF during right atrial electrophysiological stimulation to levels observed for AngII-naive animals. Left atrial epicardial mapping studies demonstrated preservation of conduction homogeneity by OA-NO2. The protection from fibrotic remodelling was mediated by suppression of Smad2-dependent myofibroblast transdifferentiation and inhibition of Nox2-dependent atrial superoxide formation.
Conclusion: OA-NO2 potently inhibits atrial fibrosis and subsequent AF. Nitro-fatty acids and possibly other lipid electrophiles thus emerge as potential therapeutic agents for AF, either by increasing endogenous levels through dietary modulation or by administration as synthetic drugs.
Keywords: Atrial fibrillation; Fibrosis; Nitro-fatty acids; Reactive oxygen species.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2015. For permissions please email: journals.permissions@oup.com.
Figures
References
-
- Camm AJ, Kirchhof P, Lip GY, Schotten U, Savelieva I, Ernst S, Van Gelder IC, Al-Attar N, Hindricks G, Prendergast B, Heidbuchel H, Alfieri O, Angelini A, Atar D, Colonna P, De Caterina R, De Sutter J, Goette A, Gorenek B, Heldal M, Hohloser SH, Kolh P, Le Heuzey JY, Ponikowski P, Rutten FH. Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). 2010;31:2369–2429. - PubMed
-
- Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. 2008;51:802–809. - PubMed
-
- Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, Pillai R, Channon KM, Casadei B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. 2005;97:629–636. - PubMed
-
- Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. 1994;269:26066–26075. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
