

Mutant SOD1 Increases APP Expression and Phosphorylation in Cellular and Animal Models of ALS
- PMID: 26600047
- PMCID: PMC4658003
- DOI: 10.1371/journal.pone.0143420
Mutant SOD1 Increases APP Expression and Phosphorylation in Cellular and Animal Models of ALS
Abstract
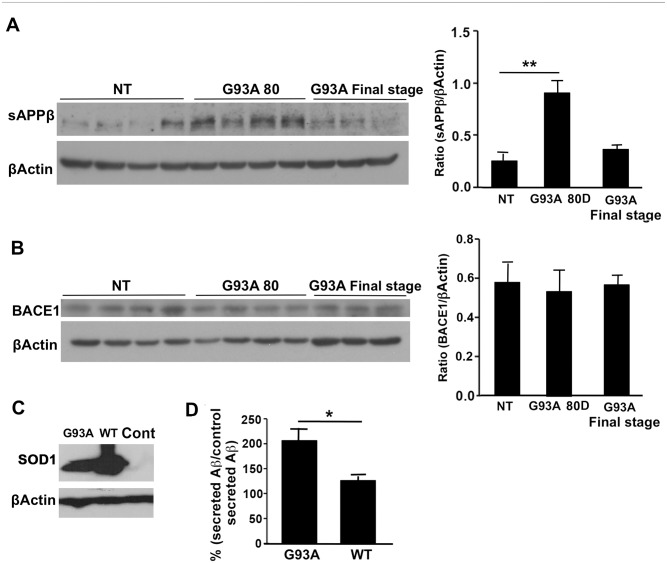
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease and it is the most common adult onset neurodegenerative disorder affecting motor neurons. There is currently no effective treatment for ALS and our understanding of the pathological mechanism is still far away from prevention and/or treatment of this devastating disease. Amyloid precursor protein (APP) is a transmembrane protein that undergoes processing either by β-secretase or α-secretase, followed by γ-secretase. In the present study, we show that APP levels, and aberrant phosphorylation, which is associated with enhanced β-secretase cleavage, are increased in SOD1G93A ALS mouse model. Fluorescence resonance energy transfer (FRET) analysis suggests a close interaction between SOD1 and APP at hippocampal synapses. Notably, SOD1G93A mutation induces APP-SOD1 conformational changes, indicating a crosstalk between these two signaling proteins. Inhibition of APP processing via monoclonal antibody called BBS that blocks APP β-secretase cleavage site, resulted in reduction of mutant SOD1G93A levels in animal and cellular models of ALS, significantly prolonged life span of SOD1G93A mice and diminished inflammation. Beyond its effect on toxic mutant SOD1G93A, BBS treatment resulted in a reduction in the levels of APP, its processing product soluble APPβ and pro-apoptotic p53. This study demonstrates that APP and its processing products contribute to ALS pathology through several different pathways; thus BBS antibody could be a promising neuroprotective strategy for treatment of this disease.
Conflict of interest statement
Figures





References
-
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264: 1772–5. Available: http://www.ncbi.nlm.nih.gov/pubmed/8209258. - PubMed
-
- Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J Biol Chem. 1994;269: 2637–44. Available: http://www.ncbi.nlm.nih.gov/pubmed/8300594. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
