

Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model
- PMID: 26601156
- PMCID: PMC4643829
- DOI: 10.1126/sciadv.1500078
Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model
Abstract
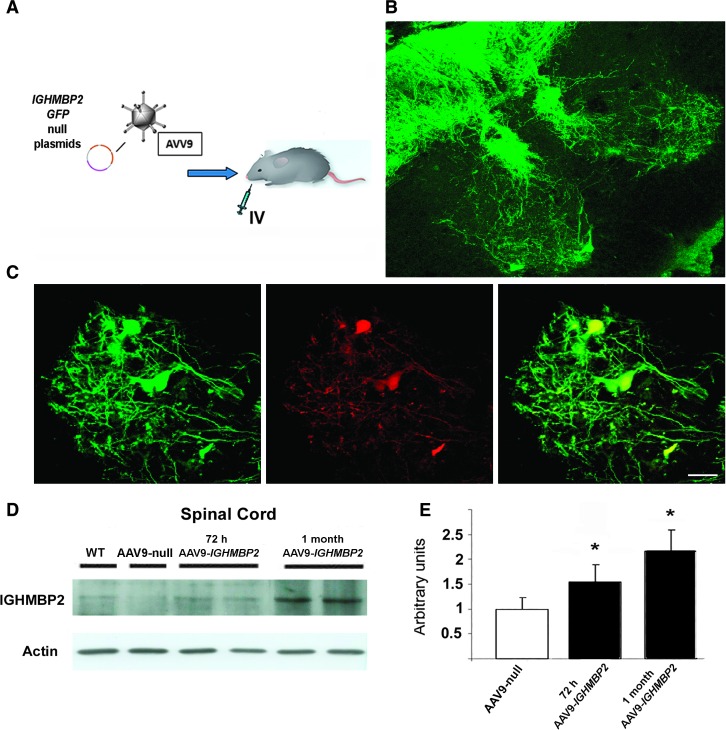
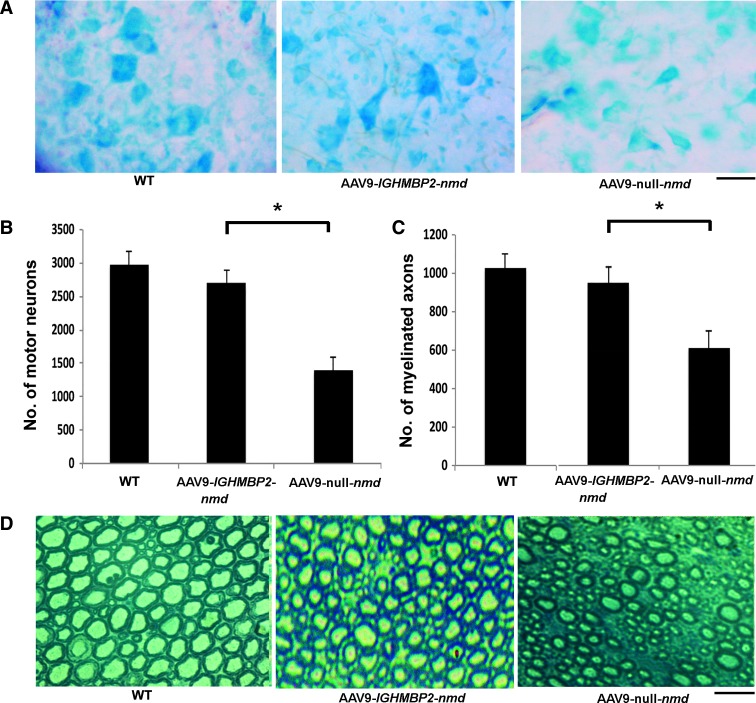
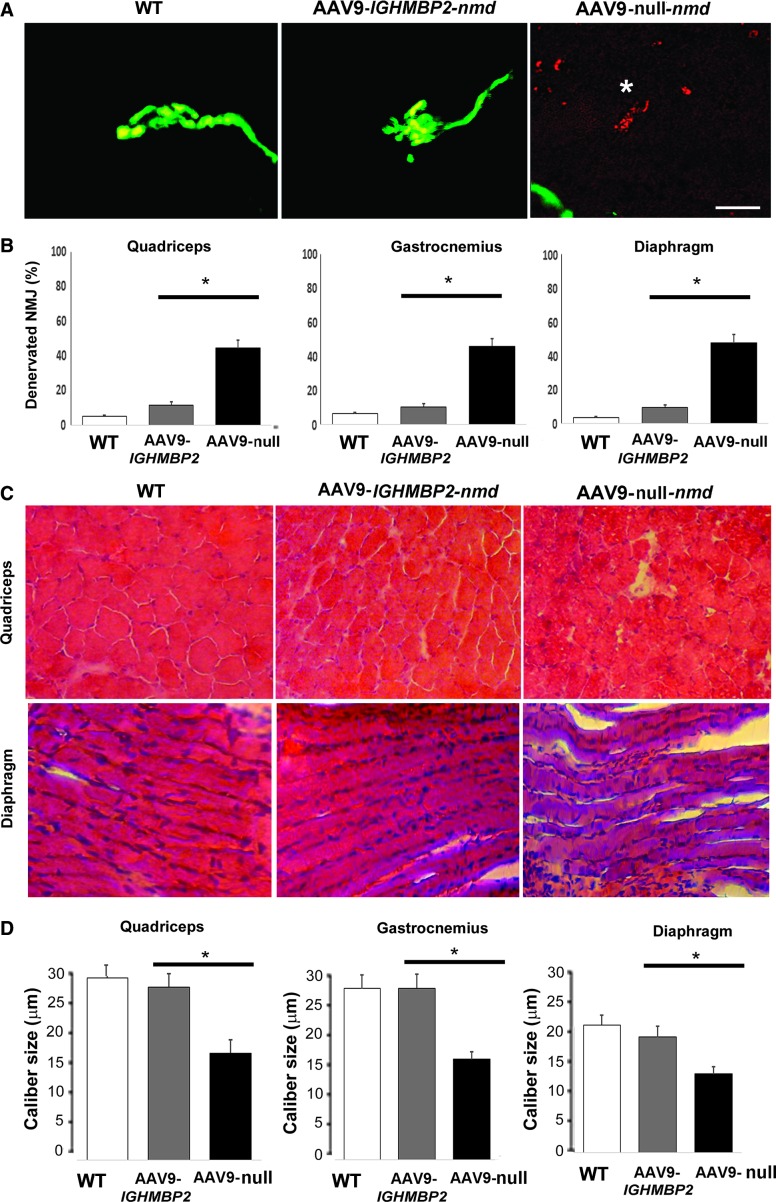
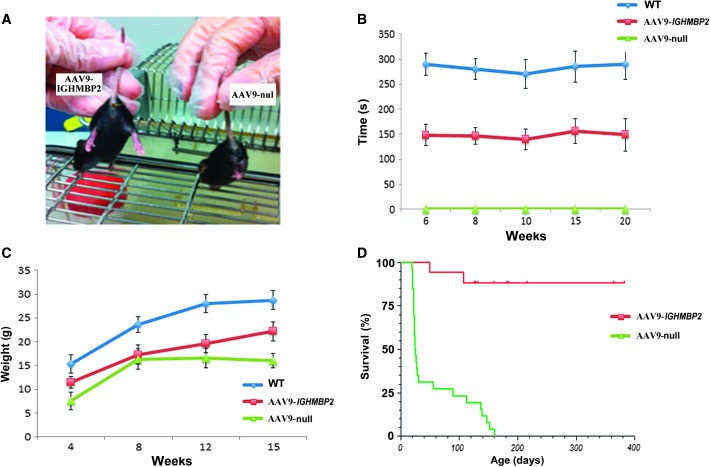
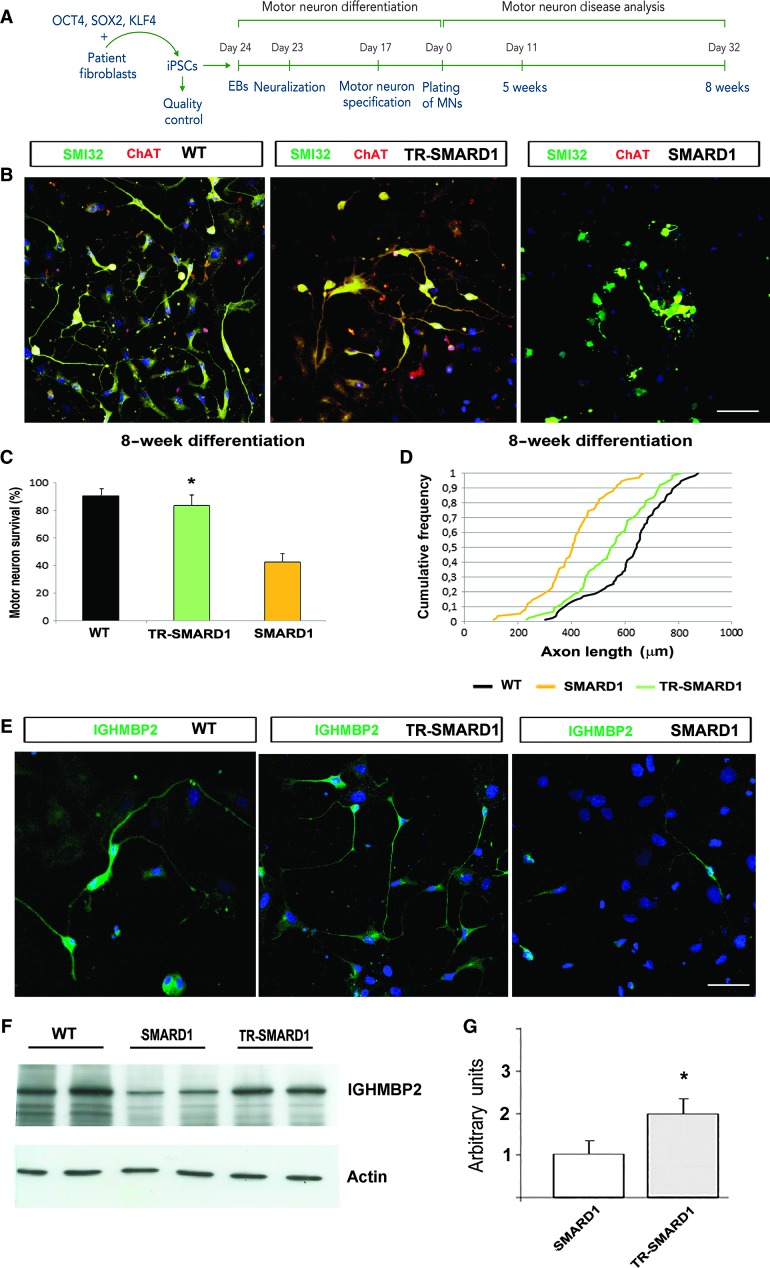
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is an autosomal recessive motor neuron disease affecting children. It is caused by mutations in the IGHMBP2 gene (11q13) and presently has no cure. Recently, adeno-associated virus serotype 9 (AAV9)-mediated gene therapy has been shown to rescue the phenotype of animal models of another lower motor neuron disorder, spinal muscular atrophy 5q, and a clinical trial with this strategy is ongoing. We report rescue of the disease phenotype in a SMARD1 mouse model after therapeutic delivery via systemic injection of an AAV9 construct encoding the wild-type IGHMBP2 to replace the defective gene. AAV9-IGHMBP2 administration restored protein levels and rescued motor function, neuromuscular physiology, and life span (450% increase), ameliorating pathological features in the central nervous system, muscles, and heart. To test this strategy in a human model, we transferred wild-type IGHMBP2 into human SMARD1-induced pluripotent stem cell-derived motor neurons; these cells exhibited increased survival and axonal length in long-term culture. Our data support the translational potential of AAV-mediated gene therapies for SMARD1, opening the door for AAV9-mediated therapy in human clinical trials.
Keywords: Spinal Muscular atrophy with Respiratory Distress Type 1; gene therapy.
Figures
References
-
- Grohmann K., Schuelke M., Diers A., Hoffmann K., Lucke B., Adams C., Bertini E., Leonhardt-Horti H., Muntoni F., Ouvrier R., Pfeufer A., Rossi R., Van Maldergem L., Wilmshurst J. M., Wienker T. F., Sendtner M., Rudnik-Schöneborn S., Zerres K., Hübner C., Mutations in the gene encoding immunoglobulin μ-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 29, 75–77 (2001). - PubMed
-
- Eckart M., Guenther U. P., Idkowiak J., Varon R., Grolle B., Boffi P., Van Maldergem L., Hübner C., Schuelke M., von Au K., The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics 129, e148–e156 (2012). - PubMed
-
- Porro F., Rinchetti P., Magri F., Riboldi G., Nizzardo M., Simone C., Zanetta C., Faravelli I., Corti S., The wide spectrum of clinical phenotypes of spinal muscular atrophy with respiratory distress type 1: A systematic review. J. Neurol. Sci. 346, 35–42 (2014). - PubMed
-
- Guenther U. P., Handoko L., Laggerbauer B., Jablonka S., Chari A., Alzheimer M., Ohmer J., Plöttner O., Gehring N., Sickmann A., von Au K., Schuelke M., Fischer U., IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1). Hum. Mol. Genet. 18, 1288–1300 (2009). - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
