

Elevations of C14:1 and C14:2 Plasma Acylcarnitines in Fasted Children: A Diagnostic Dilemma
- PMID: 26602010
- PMCID: PMC4729603
- DOI: 10.1016/j.jpeds.2015.10.045
Elevations of C14:1 and C14:2 Plasma Acylcarnitines in Fasted Children: A Diagnostic Dilemma
Abstract
Objectives: To test whether follow-up testing for very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency uncovers a diagnosis in patients with elevations of C14:1 and C14:2 plasma acylcarnitines after a controlled fasting study performed for clinically suspected hypoglycemia and to compare the acylcarnitine profiles from fasted patients without VLCAD deficiency vs patients with known VLCAD deficiency to determine whether metabolite testing distinguishes these groups.
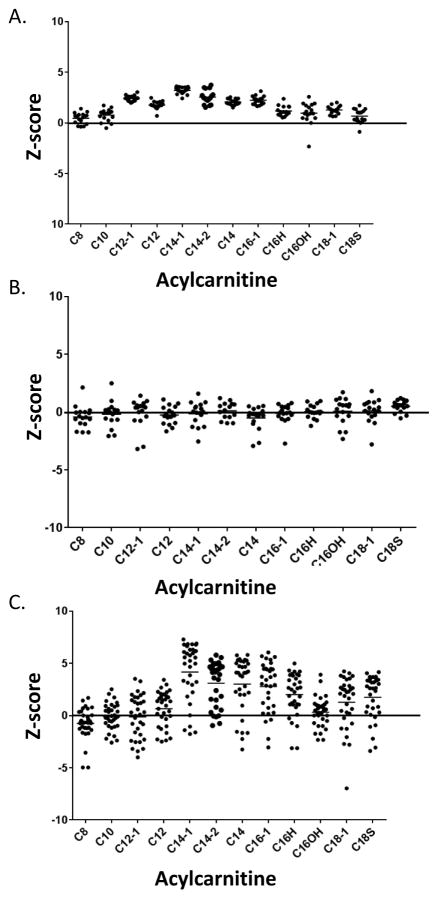
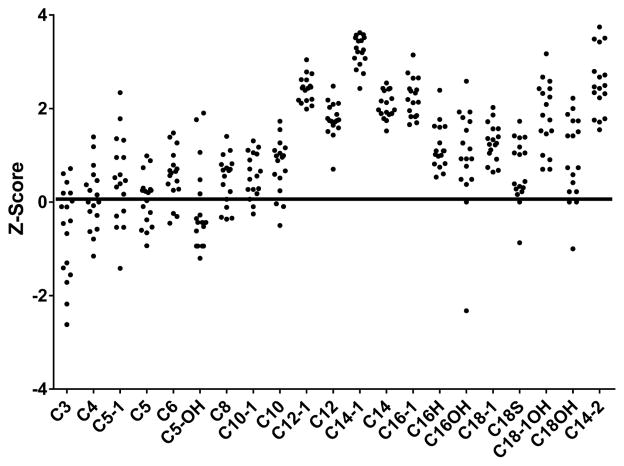
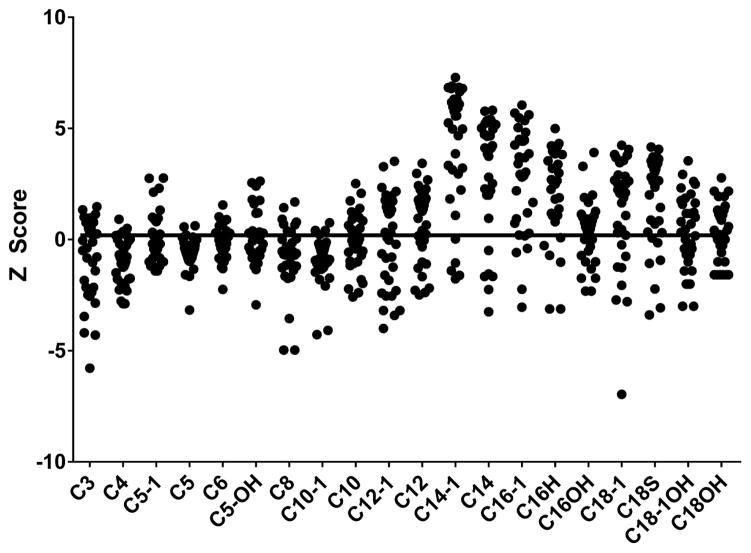
Study design: We performed a retrospective chart review and identified 17 patients with elevated C14:1 and C14:2 plasma acylcarnitine levels after a controlled fast and with testing for VLCAD deficiency (ACADVL sequencing or fibroblast fatty acid oxidation studies). The follow-up testing in all patients was inconsistent with a diagnosis of VLCAD deficiency. We compared the plasma acylcarnitine profiles from these fasted patients vs patients with VLCAD deficiency.
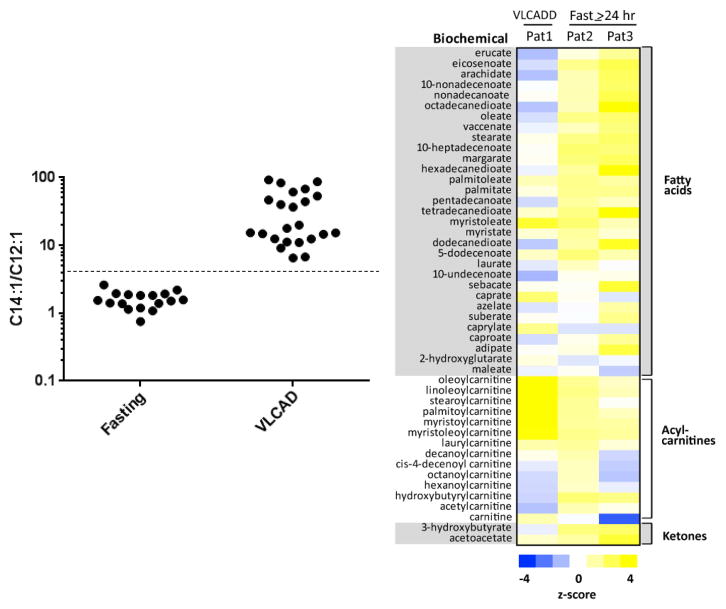
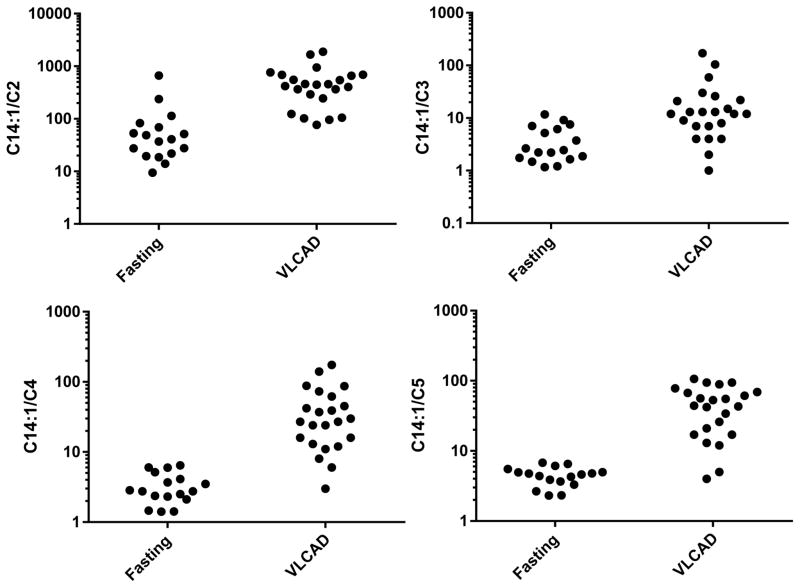
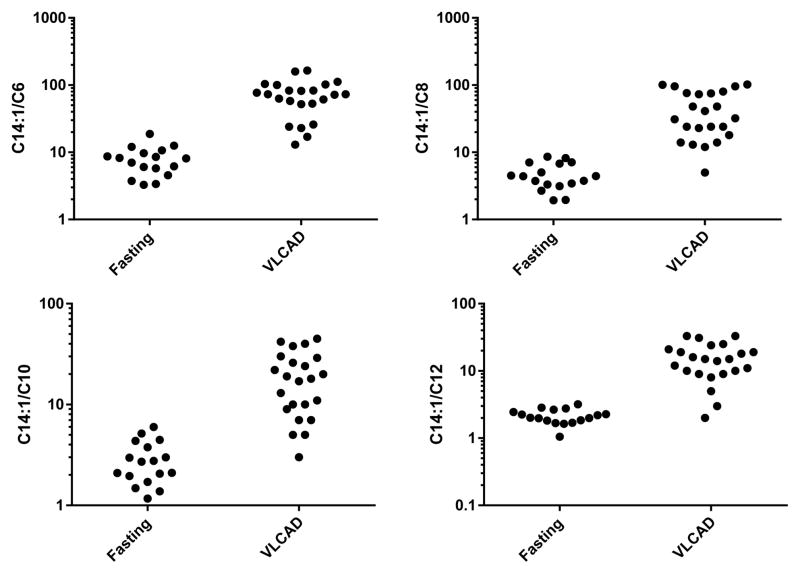
Results: C14:1/C12:1 was significantly lower (P < .001) in fasted patients vs patients with VLCAD deficiency. Metabolomics analysis performed in 2 fasted patients and 1 patient with VLCAD deficiency demonstrated evidence for up-regulated lipolysis and β-oxidation in the fasted state.
Conclusions: Elevations of plasma C14:1 and C14:2 acylcarnitines appear to be a physiologic result of lipolysis that occurs with fasting. Both metabolomics analysis and/or C14:1/C12:1 may distinguish C14:1 elevations from physiologic fasting-induced lipolysis vs VLCAD deficiency.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
A heterozygous missense mutation in adolescent-onset very long-chain acyl-CoA dehydrogenase deficiency with exercise-induced rhabdomyolysis.Tohoku J Exp Med. 2015 Apr;235(4):305-10. doi: 10.1620/tjem.235.305. Tohoku J Exp Med. 2015. PMID: 25843429
-
Newborn screening follow-up for very long-chain acyl-CoA dehydrogenase deficiency in Colorado: Working towards a standardized protocol.Mol Genet Metab. 2025 May;145(1):109104. doi: 10.1016/j.ymgme.2025.109104. Epub 2025 Apr 3. Mol Genet Metab. 2025. PMID: 40215729
-
Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database.Mol Genet Metab. 2016 Aug;118(4):272-81. doi: 10.1016/j.ymgme.2016.05.007. Epub 2016 May 13. Mol Genet Metab. 2016. PMID: 27209629 Free PMC article.
-
Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency.J Hum Genet. 2019 Feb;64(2):73-85. doi: 10.1038/s10038-018-0527-7. Epub 2018 Nov 6. J Hum Genet. 2019. PMID: 30401918 Review.
-
Very long-chain acyl-CoA dehydrogenase (VLCAD-) deficiency-studies on treatment effects and long-term outcomes in mouse models.J Inherit Metab Dis. 2017 May;40(3):317-323. doi: 10.1007/s10545-017-0016-8. Epub 2017 Feb 28. J Inherit Metab Dis. 2017. PMID: 28247148 Review.
Cited by
-
2-Pyrrolidinone and Succinimide as Clinical Screening Biomarkers for GABA-Transaminase Deficiency: Anti-seizure Medications Impact Accurate Diagnosis.Front Neurosci. 2019 May 8;13:394. doi: 10.3389/fnins.2019.00394. eCollection 2019. Front Neurosci. 2019. PMID: 31133775 Free PMC article.
-
Four novel variants identified in the ACADVL gene causing very-long-chain acyl-coenzyme A dehydrogenase deficiency in four unrelated Chinese families.Front Genet. 2024 Aug 12;15:1433160. doi: 10.3389/fgene.2024.1433160. eCollection 2024. Front Genet. 2024. PMID: 39188284 Free PMC article.
-
Biochemical Markers for the Diagnosis of Mitochondrial Fatty Acid Oxidation Diseases.J Clin Med. 2021 Oct 22;10(21):4855. doi: 10.3390/jcm10214855. J Clin Med. 2021. PMID: 34768374 Free PMC article. Review.
-
Sharpening Precision Medicine by a Thorough Interrogation of Metabolic Individuality.Comput Struct Biotechnol J. 2016 Jan 21;14:97-105. doi: 10.1016/j.csbj.2016.01.001. eCollection 2016. Comput Struct Biotechnol J. 2016. PMID: 26929792 Free PMC article. Review.
-
Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle.Rev Endocr Metab Disord. 2018 Mar;19(1):93-106. doi: 10.1007/s11154-018-9448-1. Rev Endocr Metab Disord. 2018. PMID: 29926323 Free PMC article. Review.
References
-
- Wood JC, Magera MJ, Rinaldo P, Seashore MR, Strauss AW, Friedman A. Diagnosis of very long chain acyl-dehydrogenase deficiency from an infant’s newborn screening card. Pediatrics. 2001;108:E19. - PubMed
-
- Schymik I, Liebig M, Mueller M, Wendel U, Mayatepek E, Strauss AW, et al. Pitfalls of neonatal screening for very-long-chain acyl-CoA dehydrogenase deficiency using tandem mass spectrometry. J Pediatr. 2006;149:128–30. - PubMed
-
- Boneh A, Andresen BS, Gregersen N, Ibrahim M, Tzanakos N, Peters H, et al. VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab. 2006;88:166–70. - PubMed
-
- Browning MF, Larson C, Strauss A, Marsden DL. Normal acylcarnitine levels during confirmation of abnormal newborn screening in long-chain fatty acid oxidation defects. J Inherit Metab Dis. 2005;28:545–50. - PubMed
-
- Liebig M, Schymik I, Mueller M, Wendel U, Mayatepek E, Ruiter J, et al. Neonatal screening for very long-chain acyl-coA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics. 2006;118:1065–9. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
