

A Systematic Approach to Identify Candidate Transcription Factors that Control Cell Identity
- PMID: 26603904
- PMCID: PMC4649293
- DOI: 10.1016/j.stemcr.2015.09.016
A Systematic Approach to Identify Candidate Transcription Factors that Control Cell Identity
Abstract
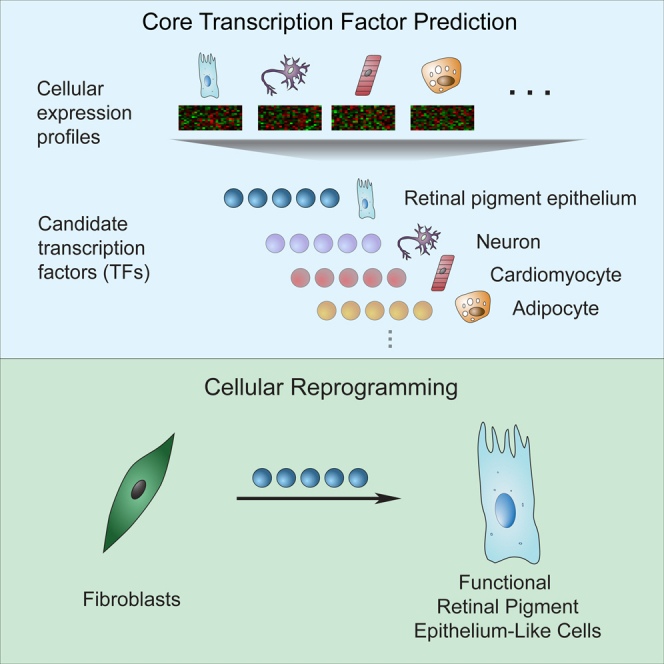
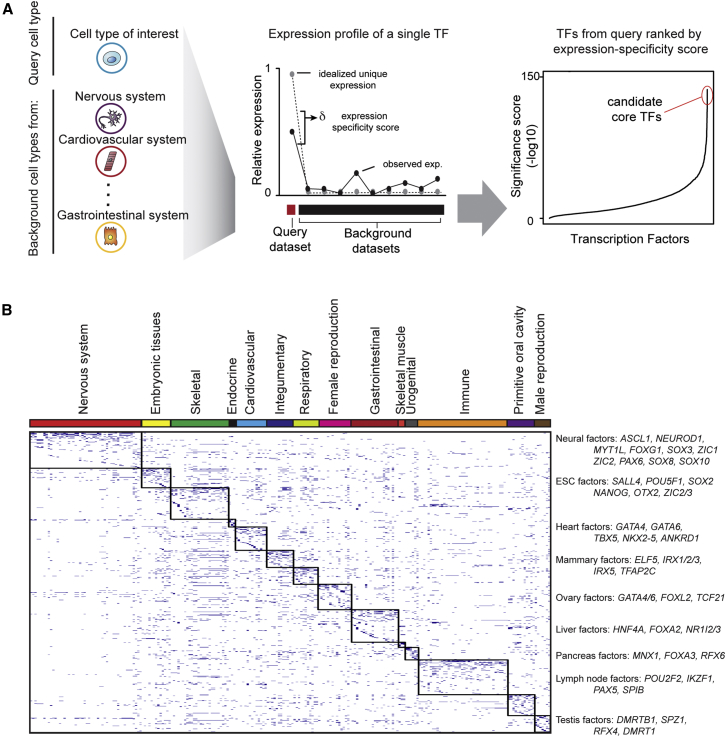
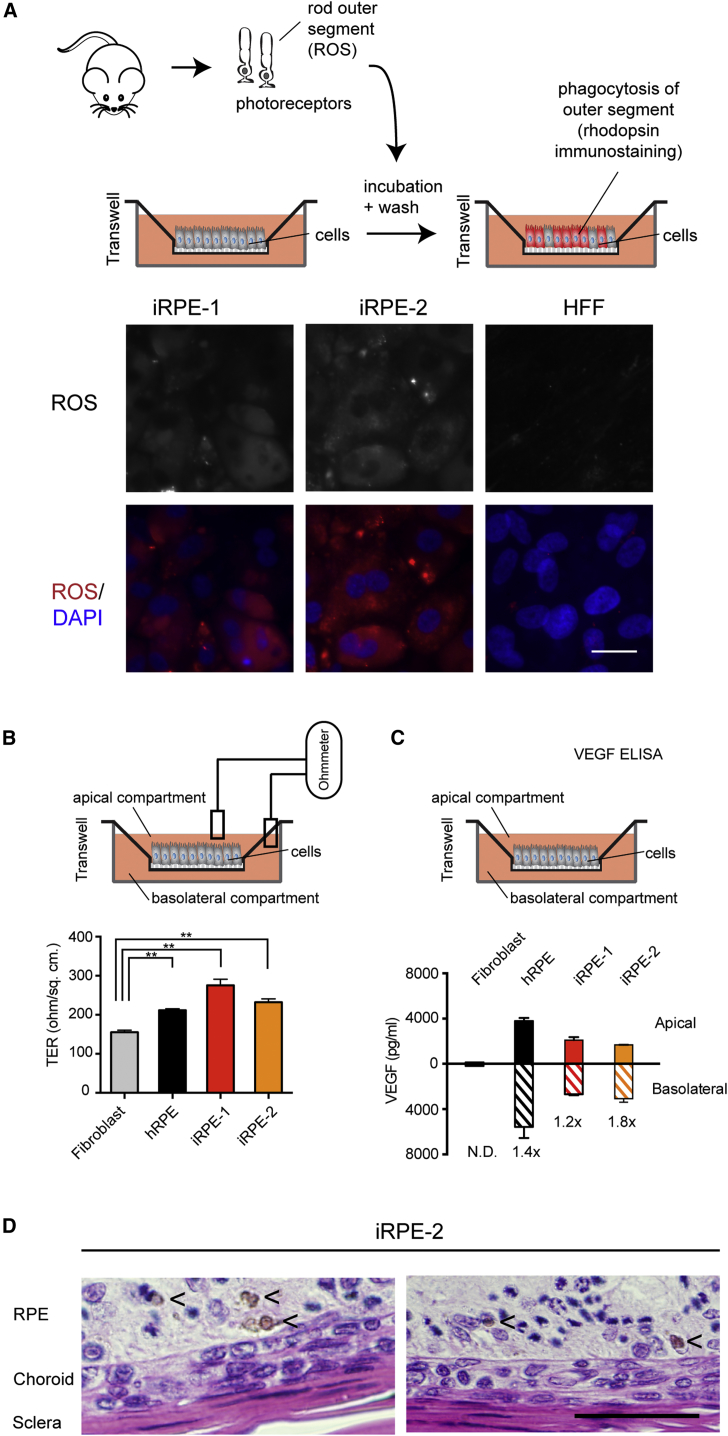
Hundreds of transcription factors (TFs) are expressed in each cell type, but cell identity can be induced through the activity of just a small number of core TFs. Systematic identification of these core TFs for a wide variety of cell types is currently lacking and would establish a foundation for understanding the transcriptional control of cell identity in development, disease, and cell-based therapy. Here, we describe a computational approach that generates an atlas of candidate core TFs for a broad spectrum of human cells. The potential impact of the atlas was demonstrated via cellular reprogramming efforts where candidate core TFs proved capable of converting human fibroblasts to retinal pigment epithelial-like cells. These results suggest that candidate core TFs from the atlas will prove a useful starting point for studying transcriptional control of cell identity and reprogramming in many human cell types.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures




References
-
- Bok D. The retinal pigment epithelium: a versatile partner in vision. J. Cell Sci. Suppl. 1993;17:189–195. - PubMed
-
- Breitling R., Armengaud P., Amtmann A., Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004;573:83–92. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
