

Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa
- PMID: 26604259
- PMCID: PMC4669384
- DOI: 10.1128/mBio.01796-15
Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa
Abstract
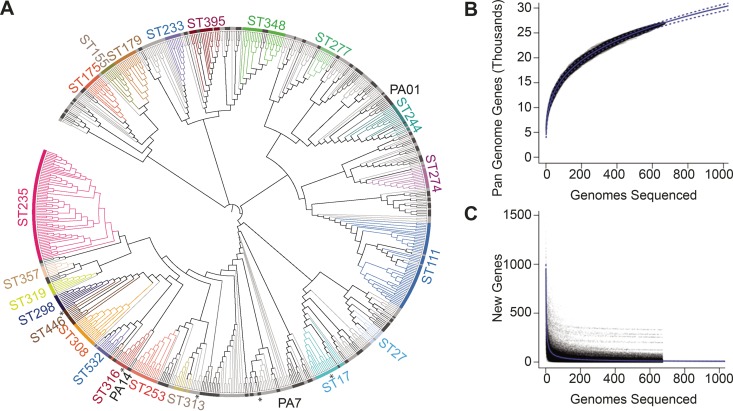
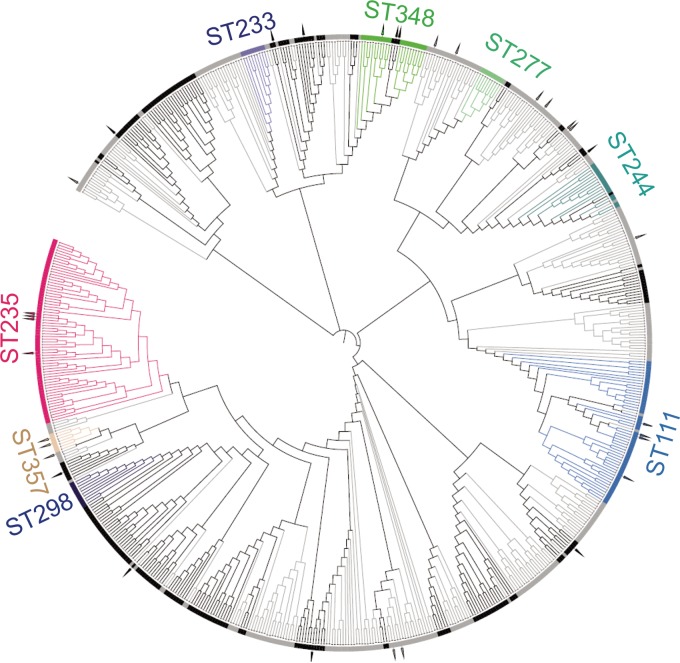
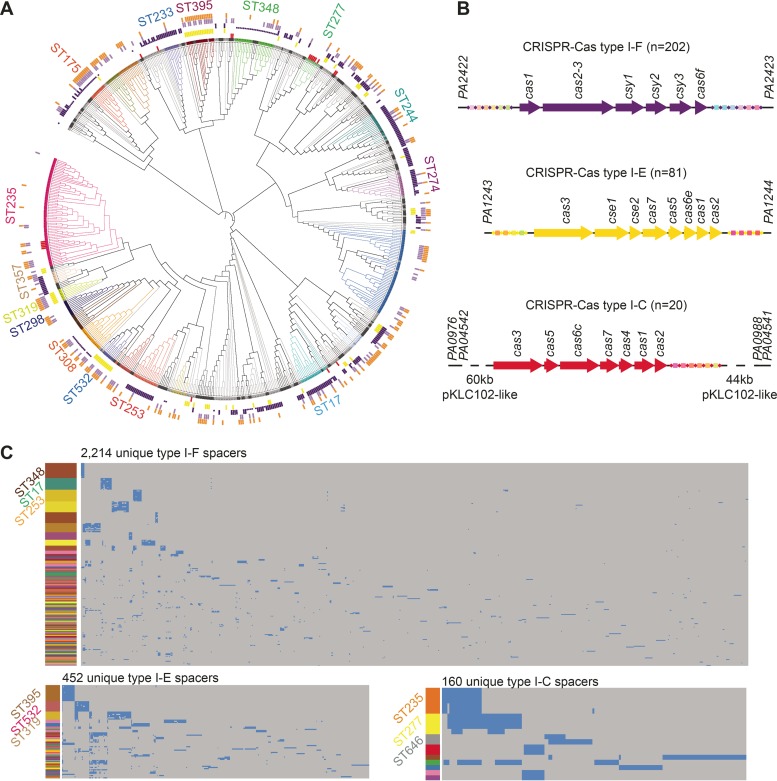
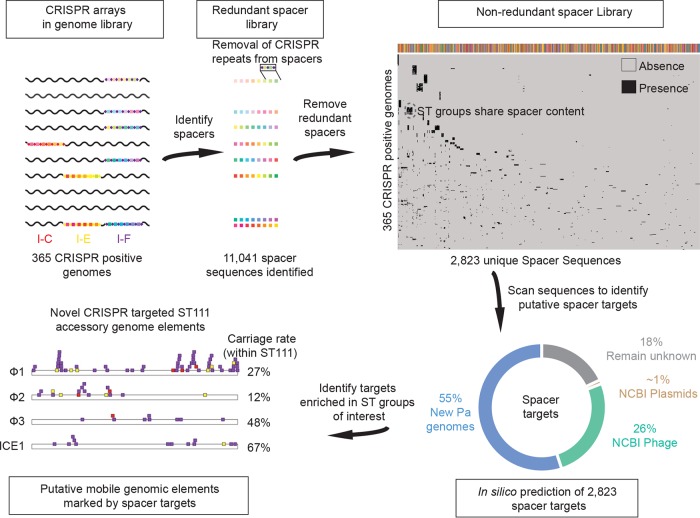
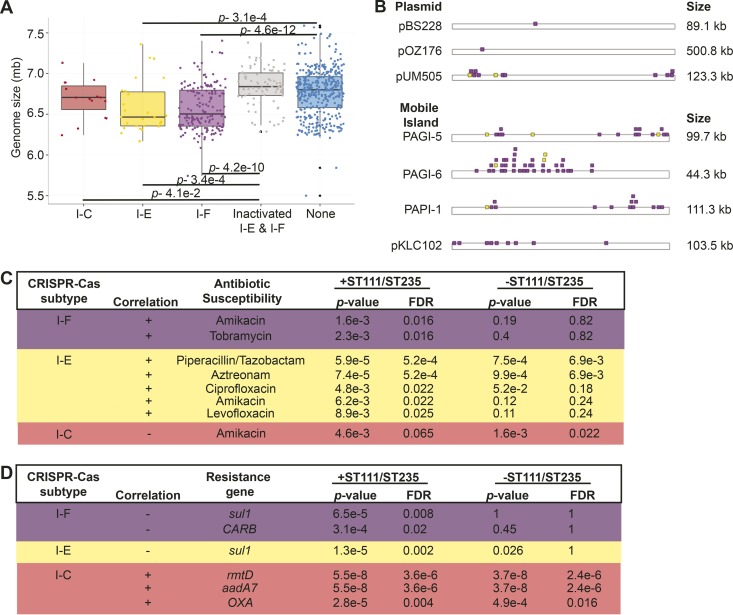
Pseudomonas aeruginosa is an antibiotic-refractory pathogen with a large genome and extensive genotypic diversity. Historically, P. aeruginosa has been a major model system for understanding the molecular mechanisms underlying type I clustered regularly interspaced short palindromic repeat (CRISPR) and CRISPR-associated protein (CRISPR-Cas)-based bacterial immune system function. However, little information on the phylogenetic distribution and potential role of these CRISPR-Cas systems in molding the P. aeruginosa accessory genome and antibiotic resistance elements is known. Computational approaches were used to identify and characterize CRISPR-Cas systems within 672 genomes, and in the process, we identified a previously unreported and putatively mobile type I-C P. aeruginosa CRISPR-Cas system. Furthermore, genomes harboring noninhibited type I-F and I-E CRISPR-Cas systems were on average ~300 kb smaller than those without a CRISPR-Cas system. In silico analysis demonstrated that the accessory genome (n = 22,036 genes) harbored the majority of identified CRISPR-Cas targets. We also assembled a global spacer library that aided the identification of difficult-to-characterize mobile genetic elements within next-generation sequencing (NGS) data and allowed CRISPR typing of a majority of P. aeruginosa strains. In summary, our analysis demonstrated that CRISPR-Cas systems play an important role in shaping the accessory genomes of globally distributed P. aeruginosa isolates.
Importance: P. aeruginosa is both an antibiotic-refractory pathogen and an important model system for type I CRISPR-Cas bacterial immune systems. By combining the genome sequences of 672 newly and previously sequenced genomes, we were able to provide a global view of the phylogenetic distribution, conservation, and potential targets of these systems. This analysis identified a new and putatively mobile P. aeruginosa CRISPR-Cas subtype, characterized the diverse distribution of known CRISPR-inhibiting genes, and provided a potential new use for CRISPR spacer libraries in accessory genome analysis. Our data demonstrated the importance of CRISPR-Cas systems in modulating the accessory genomes of globally distributed strains while also providing substantial data for subsequent genomic and experimental studies in multiple fields. Understanding why certain genotypes of P. aeruginosa are clinically prevalent and adept at horizontally acquiring virulence and antibiotic resistance elements is of major clinical and economic importance.
Copyright © 2015 van Belkum et al.
Figures
References
-
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
