

Complete nitrification by Nitrospira bacteria
- PMID: 26610024
- PMCID: PMC5152751
- DOI: 10.1038/nature16461
Complete nitrification by Nitrospira bacteria
Abstract
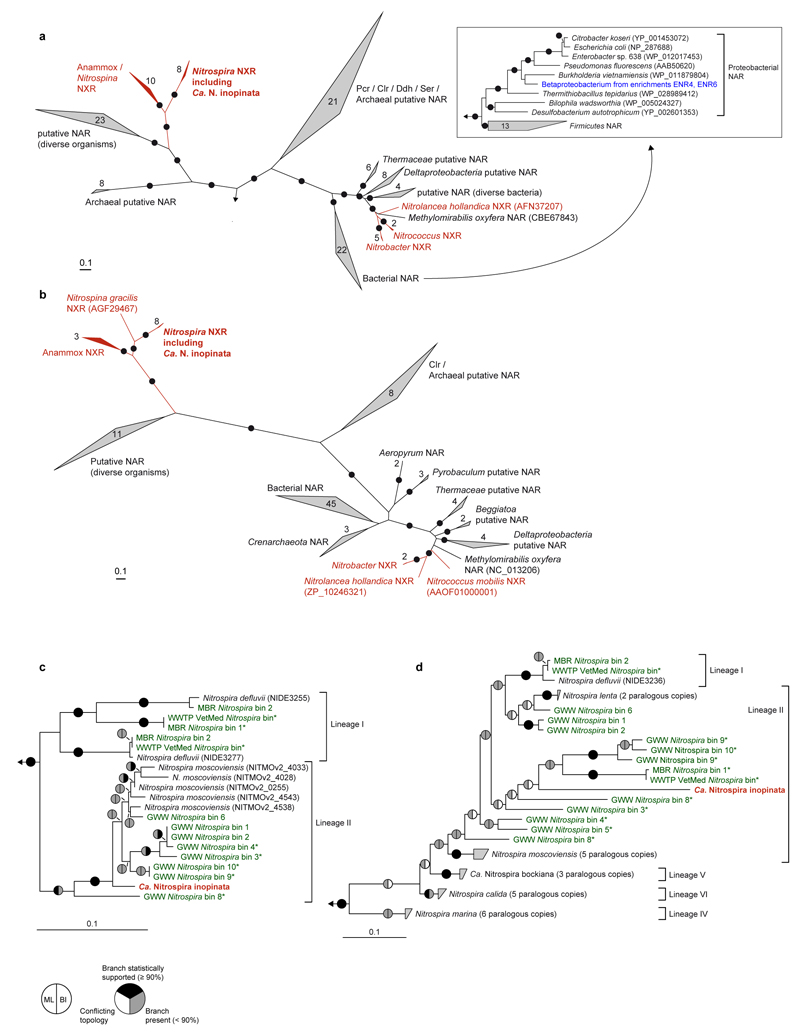
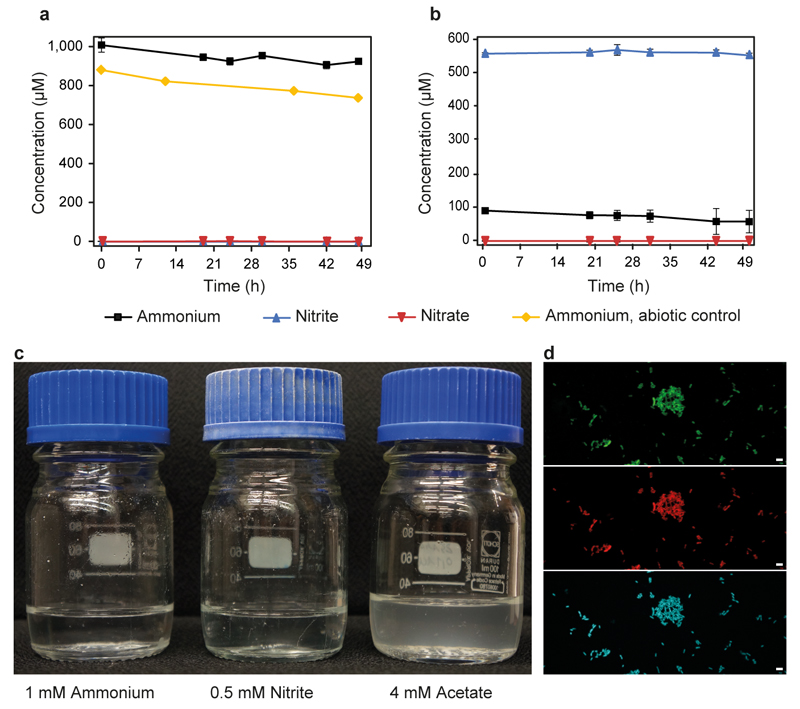
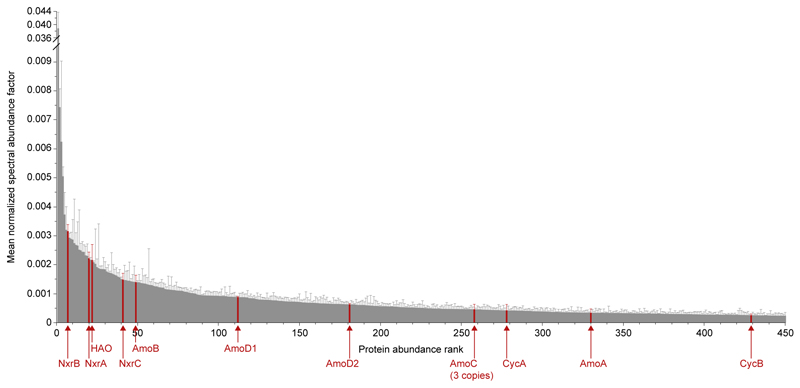
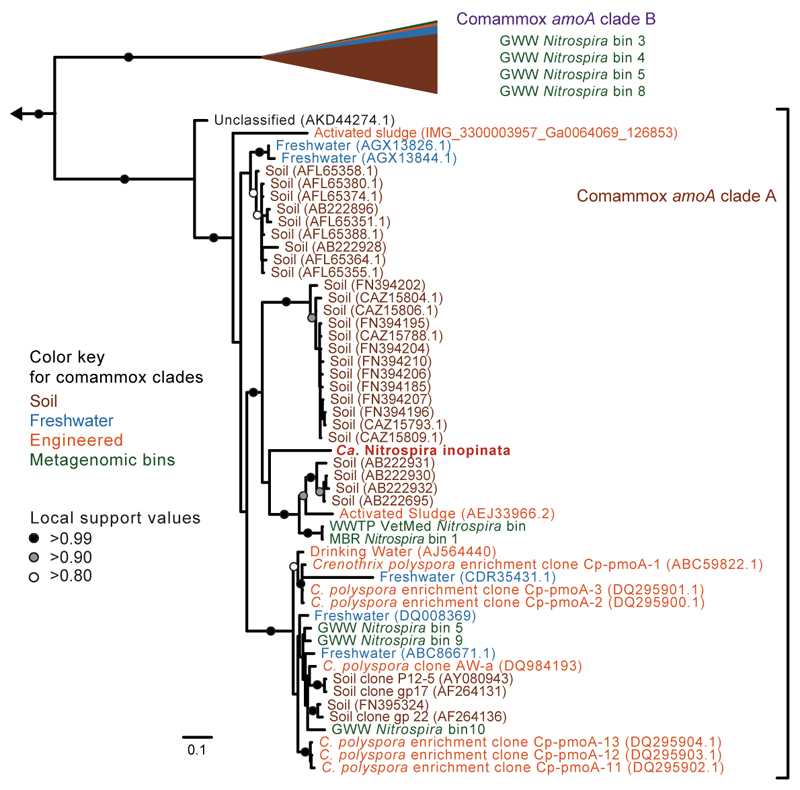
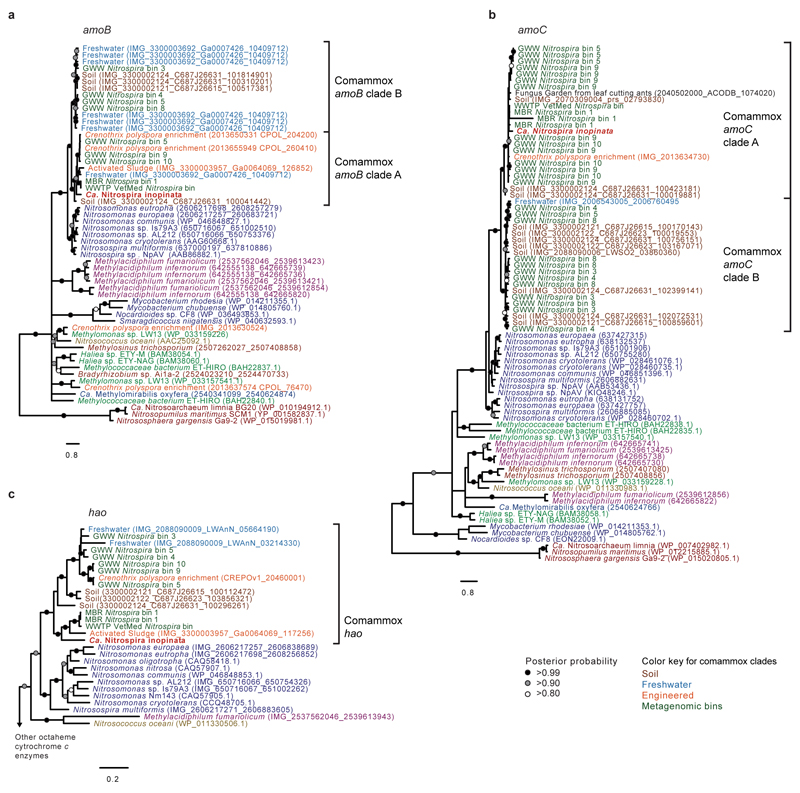
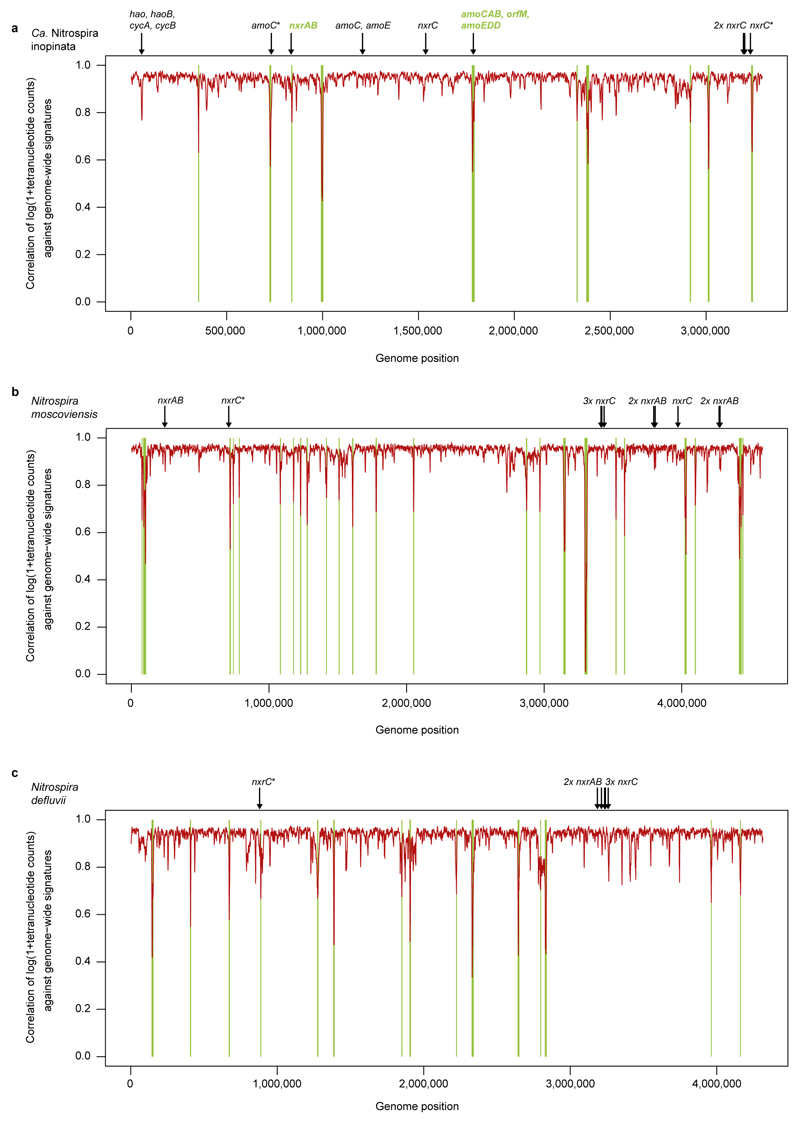
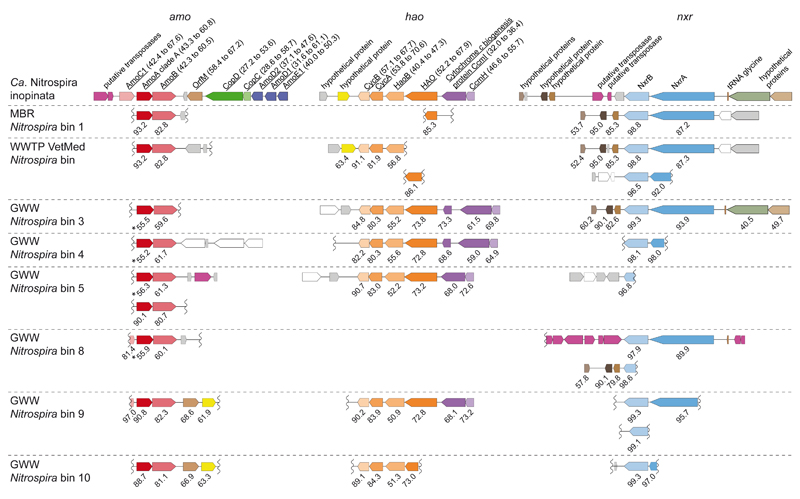
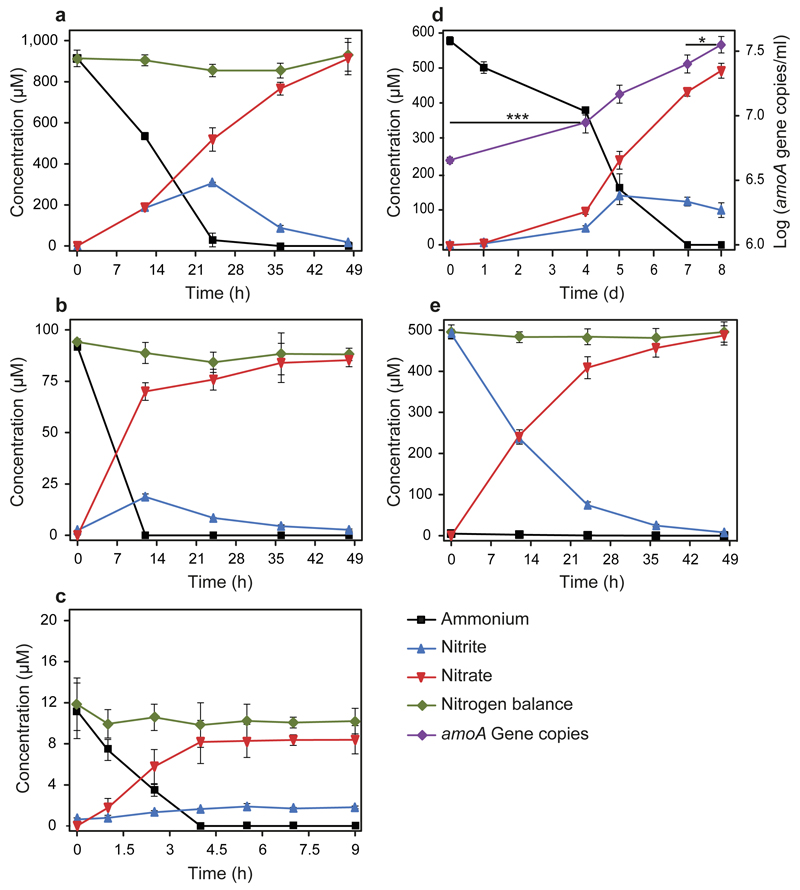
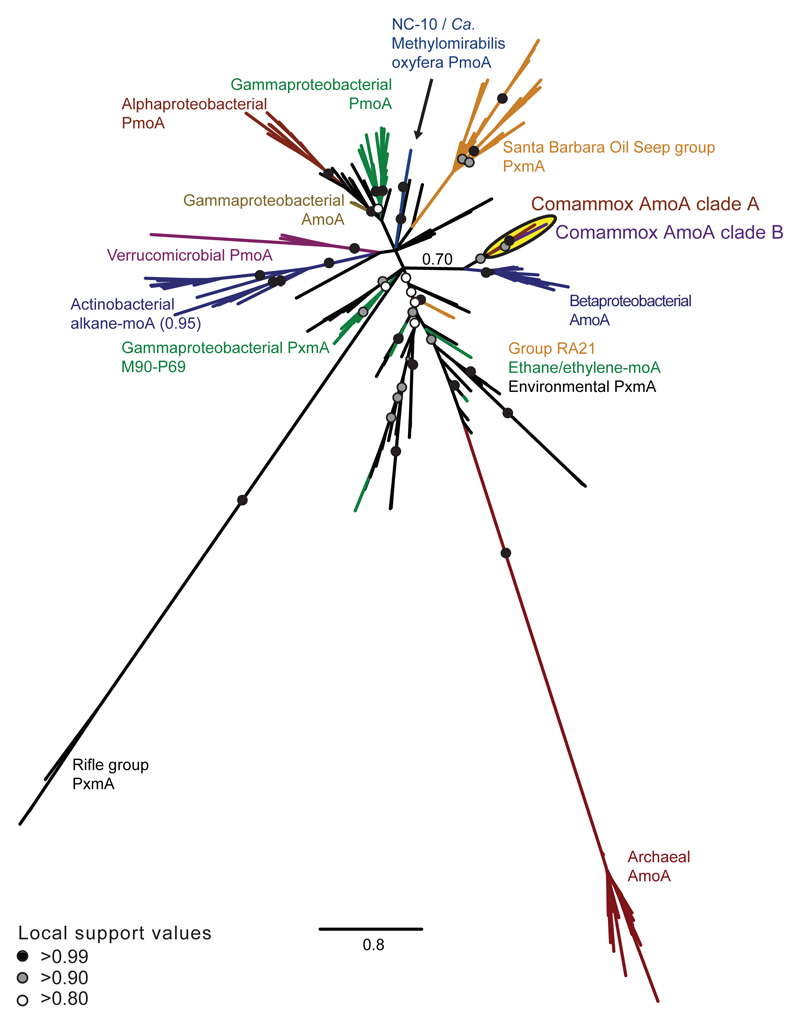
Nitrification, the oxidation of ammonia via nitrite to nitrate, has always been considered to be a two-step process catalysed by chemolithoautotrophic microorganisms oxidizing either ammonia or nitrite. No known nitrifier carries out both steps, although complete nitrification should be energetically advantageous. This functional separation has puzzled microbiologists for a century. Here we report on the discovery and cultivation of a completely nitrifying bacterium from the genus Nitrospira, a globally distributed group of nitrite oxidizers. The genome of this chemolithoautotrophic organism encodes the pathways both for ammonia and nitrite oxidation, which are concomitantly activated during growth by ammonia oxidation to nitrate. Genes affiliated with the phylogenetically distinct ammonia monooxygenase and hydroxylamine dehydrogenase genes of Nitrospira are present in many environments and were retrieved on Nitrospira-contigs in new metagenomes from engineered systems. These findings fundamentally change our picture of nitrification and point to completely nitrifying Nitrospira as key components of nitrogen-cycling microbial communities.
Figures
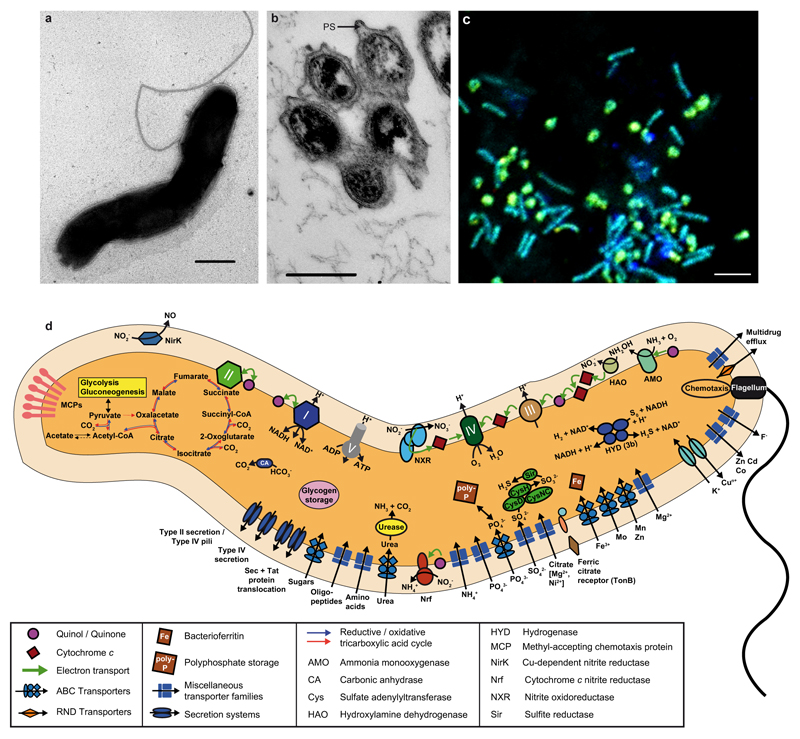
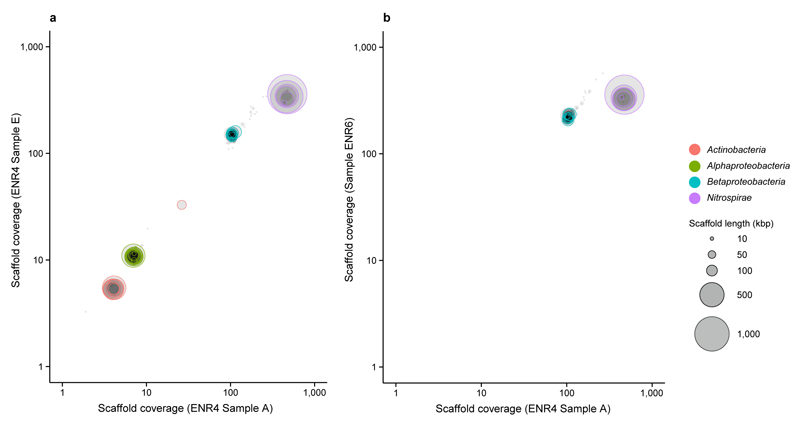
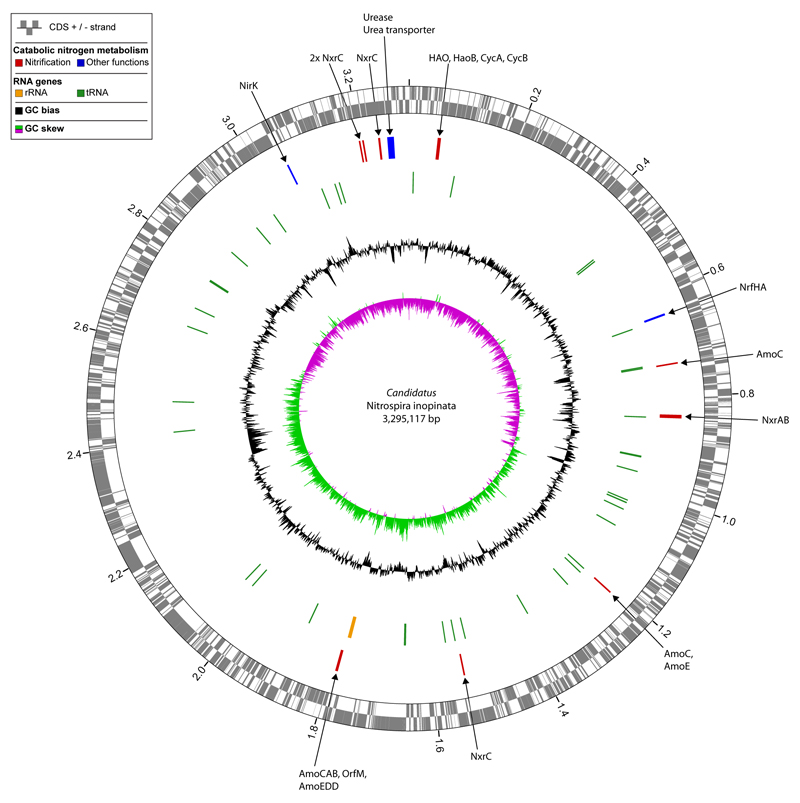
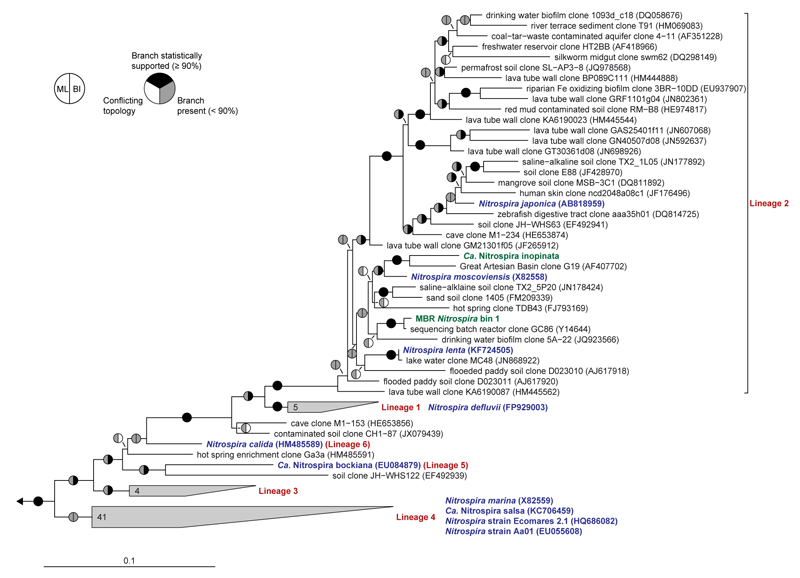
Comment in
-
Microbiology: A division of labour combined.Nature. 2015 Dec 24;528(7583):487-8. doi: 10.1038/528487a. Nature. 2015. PMID: 26701051 No abstract available.
References
-
- Bock E, Wagner M. In: The Prokaryotes: A Handbook on the Biology of Bacteria. Dworkin M, et al., editors. Springer Science+Business Media; 2001. pp. 457–495.
-
- Könneke M, et al. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature. 2005;437:543–546. - PubMed
-
- Winogradsky S. Contributions a la morphologie des organismes de la nitrification. Arch Sci Biol (St Petersb) 1892;1:88–137.
-
- Arp D, Bottomley PJ. Nitrifiers: More than 100 years from isolation to genome sequences. Microbe. 2006;1:229–234.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
