

Single-particle cryo-electron microscopy of macromolecular complexes
- PMID: 26611544
- PMCID: PMC5895108
- DOI: 10.1093/jmicro/dfv366
Single-particle cryo-electron microscopy of macromolecular complexes
Abstract
Recent technological breakthroughs in image acquisition have enabled single-particle cryo-electron microscopy (cryo-EM) to achieve near-atomic resolution structural information for biological complexes. The improvements in image quality coupled with powerful computational methods for sorting distinct particle populations now also allow the determination of compositional and conformational ensembles, thereby providing key insights into macromolecular function. However, the inherent instability and dynamic nature of biological assemblies remain a tremendous challenge that often requires tailored approaches for successful implementation of the methodology. Here, we briefly describe the fundamentals of single-particle cryo-EM with an emphasis on covering the breadth of techniques and approaches, including low- and high-resolution methods, aiming to illustrate specific steps that are crucial for obtaining structural information by this method.
Keywords: cryo-EM; macromolecular structure; negative-stain EM; single-particle EM.
© The Author 2015. Published by Oxford University Press on behalf of The Japanese Society of Microscopy. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
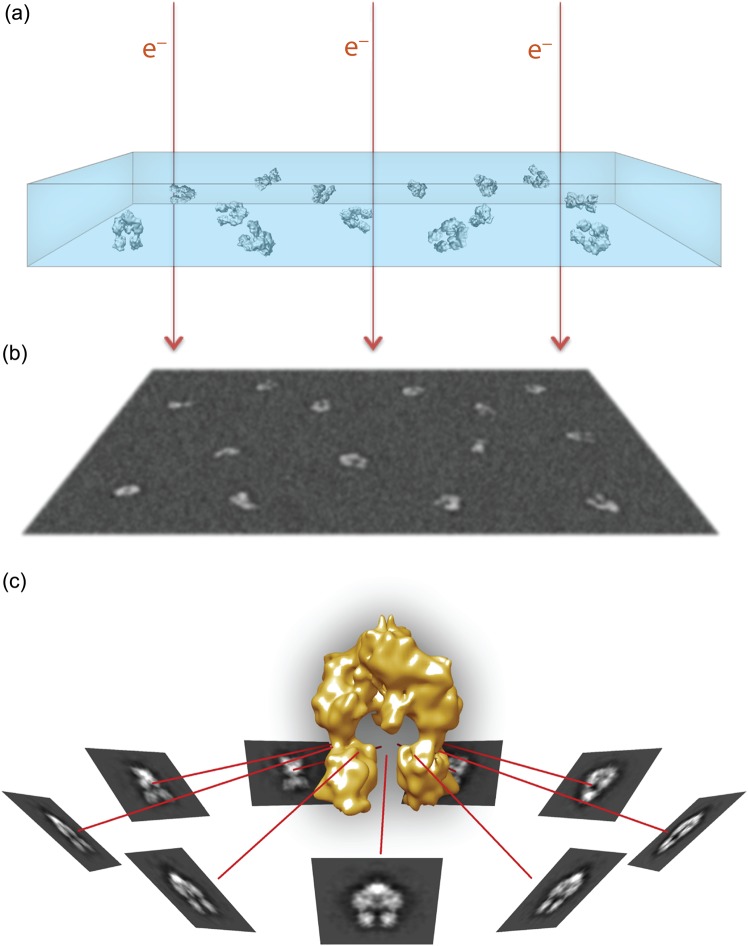
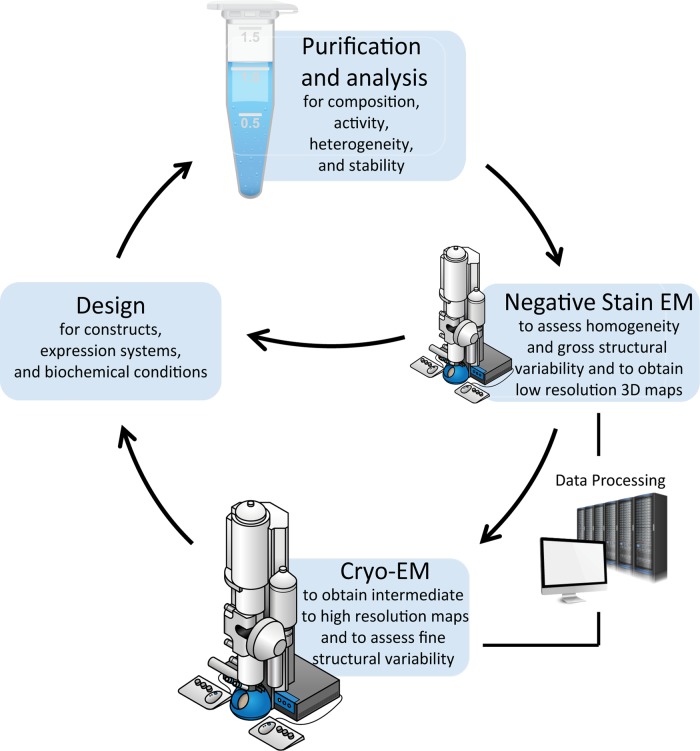
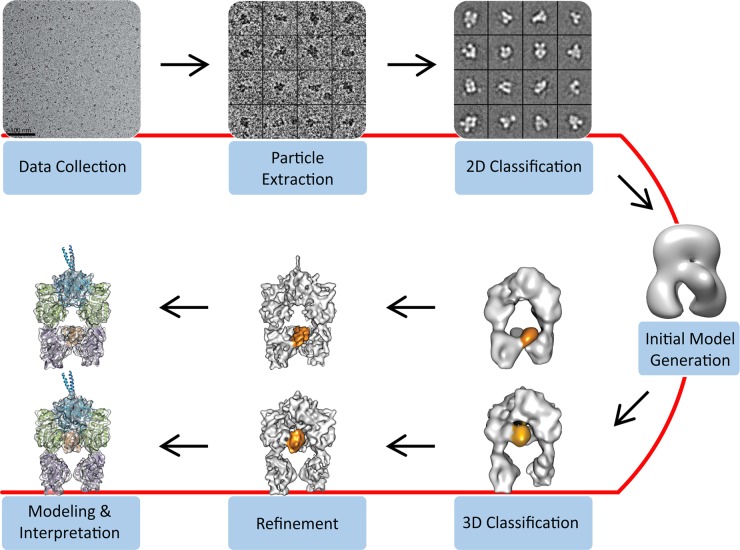
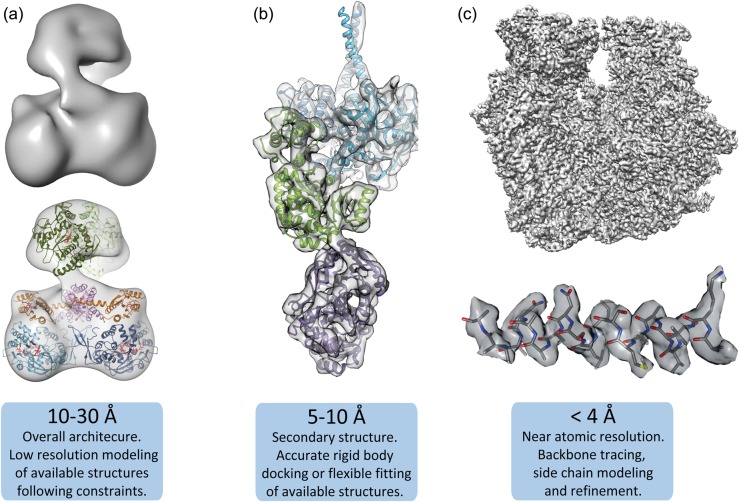
References
-
- Armache J P, Jarasch A, Anger A M, Villa E, Becker T, Bhushan S, Jossinet F, Habeck M, Dindar G, Franckenberg S, Marquez V, Mielke T, Thomm M, Berninghausen O, Beatrix B, Söding J, Westhof E, Wilson D N, Beckmann R (2010) Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-Å resolution. Proc. Natl Acad. Sci. USA 107: 19748–19753. - PMC - PubMed
-
- Ludtke S J, Baker M L, Chen D H, Song J L, Chuang D T, Chiu W (2008) De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure 16: 441–448. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
